




Abstract
Previously, we have found that phenobarbital (PB) enhanced cell survival and facilitated tumor growth in our c-myc/ transforming growth factor (TGF)-α transgenic mouse model of liver cancer. Given that PB selectively promoted initiated cells harboring β-catenin mutations during chemically induced hepatocarcinogenesis and that Wnt/β-catenin signaling is involved in both anti-apoptotic and proliferative processes, we now have extended our analysis to investigate whether promotion by PB affects the occurrence of β-catenin mutations in c-myc /TGF-α-driven tumors. The frequency of β-catenin activation as judged by somatic mutations and/or nuclear localization was significantly increased in hepatocellular carcinomas (HCCs) from c-myc /TGF-α mice treated with PB (15/28; 53.6%) as compared with that in control HCCs (2/28; 7.1%). Furthermore, an intact β-catenin locus was detected in all neoplasms following PB treatment, whereas 57.1% (16/28) of malignant tumors from c-myc /TGF-α untreated mice displayed loss of heterozygosity at the β-catenin locus. Strikingly, in the majority of PB-treated HCCs β-catenin nuclear localization was limited to small cells with high nuclear/cytoplasmic ratio forming an invasion front (NAinv). β-Catenin NAinv cells showed cytoplasmic redistribution of E-cadherin associated with intense mucin 1 and matrilysin immunostaining, suggesting their invasive phenotype. All β-catenin-positive HCCs displayed increased proliferation and tumor size, but no difference in the apoptotic rate when compared with β-catenin negative tumors. These findings show that PB treatment positively selects for a cell population displaying activation of β-catenin in c-myc /TGF-α HCCs. β-Catenin activation confers additional growth and invasive advantages in a model of liver cancer already accelerated by synergistic activity of the c-myc and TGF-α transgenes.
Introduction
Tumor promoters are non-mutagenic compounds that increase the probability of cancer development by stimulating the clonal expansion of cells transformed during tumor initiation. Phenobarbital (PB), a non-genotoxic barbiturate widely used as a hepatic tumor promoter in rodents initiated by a variety of liver carcinogens, acts on a plethora of processes that influence cell proliferation and survival ( 1 – 4 ). However, it remains unclear which PB functions are essential for tumor promotion and, as a consequence, are selected against during tumor development.
We have generated previously and extensively characterized a mouse model of liver cancer in which hepatocyte-specific co-expression of the c-myc oncogene and transforming growth factor-α (TGF-α) synergistically accelerated liver tumor growth as compared with that driven by either one of the single transgenes ( 5 , 6 ). Cooperative activity of c-myc and TGF-α promoted tumorigenicity not only by dramatically accelerating cell proliferation but also by strongly inhibiting programmed cell death ( 7 ). Interestingly, when c-myc /TGF-α mice were subjected to PB treatment, the neoplastic process was further facilitated ( 8 , 9 ). At the molecular level, PB functioned as a survival factor causing up-regulation of bcl-2 during the early stages of c-myc /TGF-α-driven tumor development ( 8 ).
Interestingly, it was found recently that selective pressure during tumor promotion by PB favors the clonal outgrowth of β-catenin-mutated tumors in a mouse model of diethylnitrosamine (DEN)-induced hepatocarcinogenesis ( 10 ). β-Catenin is a cytoskeletal component that participates in intercellular adhesion through its association with α-, γ-catenin and the E-cadherin intracellular domain ( 11 ). The cadherin/catenins interaction represents not only a prerequisite for cell–cell adhesion, but also for inhibition of cell motility and invasion by preserving epithelial tissue integrity ( 12 ). In addition to its adhesive function, β-catenin plays a crucial role in embryonic development as well as in the oncogenic transformation of many cell types ( 13 ). β-Catenin protein levels are regulated by phosphorylation of its N-terminal region by the glycogen synthase kinase-3β (GSK-3β) complex, which includes adenomatous polyposis coli (APC), axin, conductin (axin 2) and GSK-3β. Once phosphorylated, β-catenin undergoes degradation via the ubiquitin–proteasome pathway ( 14 ). Mutations in either APC or β-catenin itself ( 13 ) lead to an elevated level of free β-catenin, which translocates into the nucleus and interacts with transcription factors of the LEF/T-cell factor family. The β-catenin/T-cell factor (Tcf) complex induces the expression of Tcf-target genes involved in promotion of cell proliferation, inhibition of apoptosis and invasiveness. Tcf-target genes include c-myc , c-jun, Fra-1, Cyclin D1, Wisp and matrilysin/matrix metalloproteinase 7 ( 13 , 15 ).
Given this information and the fact that activation of the β-catenin pathway is a very rare event during c-myc /TGF-α-driven hepatocarcinogenesis ( 16 ), the present study investigates whether PB selects for cells harboring β-catenin defects during tumor promotion in the c-myc /TGF-α model of liver cancer. Our results show that activation of the β-catenin gene might in fact provide a potent proliferative and invasive advantage in a mouse model of accelerated liver carcinogenesis.
Materials and methods
Transgenic animals and treatment
Alb/ c-myc /MT/TGF-α ( c-myc /TGF-α) double-transgenic mice were generated as described elsewhere ( 5 ). Tumor samples were obtained from a previous experiment ( 8 , 9 ). In brief, at the time of weaning, male c-myc /TGF-α mice were divided into two groups: one group was fed a NIH-31-based chow (untreated mice), while the other group was maintained on the same diet containing 0.05% PB (PB-treated mice) for 28 weeks. Animal housing and care were carried out according to the National Institutes of Health guidelines.
Tissue specimens, histopathological analysis and DNA extraction
Ten percent formalin-fixed, paraffin-embedded liver sections (stained with H&E) were used for histopathological diagnoses based upon criteria described by Frith et al . ( 17 ). Neoplastic lesions were microdissected from parallel-unstained slides and DNA was extracted as described previously ( 16 ).
β-Catenin mutation and deletion analysis, DNA sequencing
PCR reactions were performed on an Eppendorf Mastercycler Gradient (Westbury, NY) using genomic DNA with BCAT-1F and BCAT-2R primers, which amplify the putative GSK-3β phosphorylation sites in β-catenin exon 2, homologous to human exon 3 ( 18 ). The PCR was carried out in a reaction volume of 50 µl consisting of 1.0 µM each primer, 0.2 mM each dNTP, 10× Cloned Pfu DNA polymerase reaction buffer, 2.5 U Pfu Turbo DNA polymerase (Stratagene, La Jolla, CA) and 0.4 µg of template DNA. Cycling parameters were: initial step of denaturation at 94°C for 2 min, followed by 35 cycles at 94°C for 30 s, 53°C for 30 s and 72°C for 30 s. The presence of large exon 2 interstitial deletions was assessed by PCR reaction using the LF and LR primers that span the whole region of the β-catenin gene ( 10 ). PCR products electrophoresed, excised and purified from 2% TAE gels with the QIAEX II gel extraction kit (Qiagen, Valencia, CA) were sequenced with the ABI PRISM BigDye Terminator Cycle Sequencing Ready Reaction Kit (Applied Biosystems, Foster City, CA).
Immunohistochemistry
Immunohistochemical stainings were performed on 10% formalin-fixed, paraffin-embedded tissues. Deparaffinized sections were incubated in 3% H 2 O 2 dissolved in PBS 1× for 30 min and microwaved in 0.001 M EDTA (pH 8.0) or 10 mM citrate buffer (pH 6.0) for 12 min. Mouse monoclonal anti-β-catenin, anti-E-cadherin (Transduction Laboratories, Lexington, KY), anti-proliferating cell nuclear antigen (PCNA) (Santa Cruz Biotechnology, Santa Cruz, CA), goat polyclonal anti-conductin, anti-axin and anti-matrix metalloproteinase 7, rabbit polyclonal anti-APC and anti-mucin 1 (MUC1) antibodies (Santa Cruz Biotechnology) were applied in a 1:100 dilution. Immunoreactivity was visualized with the Vectastain Elite ABC kit (Vector Laboratories) and 3,3′ DAB (Dako, Carpinteria, CA) as the chromogen. Immunostaining with rat monoclonal A6 antibody recognizing common surface-exposed antigens of mouse biliary epithelial and oval cells was performed as described previously ( 19 ). As negative controls, the sections were immunostained omitting the primary antibody. Specificity of immunostaining for conductin, axin, APC, MUC1 and matrilysin was determined by pre-incubating primary antibodies with respective blocking peptides as recommended by the manufacturer (Santa Cruz Biotechnology). Slides were counterstained with Gill's hematoxylin. Results of immunohistochemical findings are summarized in Table I .
Summary of DNA sequencing, LOH at the β-catenin locus and immunohistochemical findings in HCCs from c-myc /TGF-α transgenic mice
| Feature | No treatment ( n = 28) frequency (%) | Phenobarbital treatment ( n = 28) frequency (%) |
|---|---|---|
| β-Catenin mutations | 7.1 | 32.1 |
| Mutated codons | TCC→TCT (Ser 45→Phe) | TCC→TCT (Ser 45→Phe) |
| TCC→TCT (Ser 45→Phe) | TCC→TCT (Ser 45→Phe) | |
| TCC→TCT (Ser 45→Phe) | ||
| TCC→TTC (Ser 45→Phe) | ||
| TCC→TTC (Ser 45→Phe) | ||
| TCC→TTC (Ser 45→Phe) | ||
| TCC→TTC (Ser 45→Phe) | ||
| TCT→TAT (Ser33→Tyr) | ||
| TCT→TAT (Ser33→Tyr) | ||
| LOH at β-catenin locus | 57.1 | 0 |
| β-Catenin nuclear accumulation | 7.1 | 53.6 |
| β-Catenin NAd/s | 7.1 | 14.3 |
| β-Catenin NAinv | 0 | 39.3 |
| E-Cadherin cytoplasmic localization | 0 | 39.3 |
| MUC1 expression | 0 | 39.3 |
| MMP-7 expression | 0 | 39.3 |
| Feature | No treatment ( n = 28) frequency (%) | Phenobarbital treatment ( n = 28) frequency (%) |
|---|---|---|
| β-Catenin mutations | 7.1 | 32.1 |
| Mutated codons | TCC→TCT (Ser 45→Phe) | TCC→TCT (Ser 45→Phe) |
| TCC→TCT (Ser 45→Phe) | TCC→TCT (Ser 45→Phe) | |
| TCC→TCT (Ser 45→Phe) | ||
| TCC→TTC (Ser 45→Phe) | ||
| TCC→TTC (Ser 45→Phe) | ||
| TCC→TTC (Ser 45→Phe) | ||
| TCC→TTC (Ser 45→Phe) | ||
| TCT→TAT (Ser33→Tyr) | ||
| TCT→TAT (Ser33→Tyr) | ||
| LOH at β-catenin locus | 57.1 | 0 |
| β-Catenin nuclear accumulation | 7.1 | 53.6 |
| β-Catenin NAd/s | 7.1 | 14.3 |
| β-Catenin NAinv | 0 | 39.3 |
| E-Cadherin cytoplasmic localization | 0 | 39.3 |
| MUC1 expression | 0 | 39.3 |
| MMP-7 expression | 0 | 39.3 |
Summary of DNA sequencing, LOH at the β-catenin locus and immunohistochemical findings in HCCs from c-myc /TGF-α transgenic mice
| Feature | No treatment ( n = 28) frequency (%) | Phenobarbital treatment ( n = 28) frequency (%) |
|---|---|---|
| β-Catenin mutations | 7.1 | 32.1 |
| Mutated codons | TCC→TCT (Ser 45→Phe) | TCC→TCT (Ser 45→Phe) |
| TCC→TCT (Ser 45→Phe) | TCC→TCT (Ser 45→Phe) | |
| TCC→TCT (Ser 45→Phe) | ||
| TCC→TTC (Ser 45→Phe) | ||
| TCC→TTC (Ser 45→Phe) | ||
| TCC→TTC (Ser 45→Phe) | ||
| TCC→TTC (Ser 45→Phe) | ||
| TCT→TAT (Ser33→Tyr) | ||
| TCT→TAT (Ser33→Tyr) | ||
| LOH at β-catenin locus | 57.1 | 0 |
| β-Catenin nuclear accumulation | 7.1 | 53.6 |
| β-Catenin NAd/s | 7.1 | 14.3 |
| β-Catenin NAinv | 0 | 39.3 |
| E-Cadherin cytoplasmic localization | 0 | 39.3 |
| MUC1 expression | 0 | 39.3 |
| MMP-7 expression | 0 | 39.3 |
| Feature | No treatment ( n = 28) frequency (%) | Phenobarbital treatment ( n = 28) frequency (%) |
|---|---|---|
| β-Catenin mutations | 7.1 | 32.1 |
| Mutated codons | TCC→TCT (Ser 45→Phe) | TCC→TCT (Ser 45→Phe) |
| TCC→TCT (Ser 45→Phe) | TCC→TCT (Ser 45→Phe) | |
| TCC→TCT (Ser 45→Phe) | ||
| TCC→TTC (Ser 45→Phe) | ||
| TCC→TTC (Ser 45→Phe) | ||
| TCC→TTC (Ser 45→Phe) | ||
| TCC→TTC (Ser 45→Phe) | ||
| TCT→TAT (Ser33→Tyr) | ||
| TCT→TAT (Ser33→Tyr) | ||
| LOH at β-catenin locus | 57.1 | 0 |
| β-Catenin nuclear accumulation | 7.1 | 53.6 |
| β-Catenin NAd/s | 7.1 | 14.3 |
| β-Catenin NAinv | 0 | 39.3 |
| E-Cadherin cytoplasmic localization | 0 | 39.3 |
| MUC1 expression | 0 | 39.3 |
| MMP-7 expression | 0 | 39.3 |
Microsatellite analysis
The same samples were screened for microsatellite alterations at β-catenin (D9Mit121, D9Mit52), APC (D18Mit27, D18Mit15), axin (D17Mit55, D17Mit100) and conductin (D11Mit161) loci. Microsatellite primers were purchased from Research Genetics (Huntsville, AL). The microsatellite primer sequences and product sizes are shown in Table II . The PCR was carried out in a reaction volume of 10 µl consisting of 0.4 µM each primer, 200 µM each dNTP, 10× PCR buffer, 0.4 U of FastStart Taq DNA Polymerase (Roche Molecular Biochemicals) and 20 ng of template DNA, with an initial activation step at 95°C for 6 min, followed by 40 cycles (95°C for 30 s, 55°C for 45 s, 72°C for 1 min) and a final extension at 72°C for 10 min. After denaturation in TBE-Urea Sample Buffer (Novex), 12 µl of each mixture were loaded onto a 6% TBE-Urea Gels (Novex), electrophoresed at 170 V for 50 min and visualized by silver nitrate staining (Silver Stain Plus; Bio-Rad). Loss of heterozygosity (LOH) was recorded when a 50% or greater reduction in electrophoretic band intensity was detected.
Sequences of microsatellite primers used in this study
| Primer name | Sequence | Product size | ||
|---|---|---|---|---|
| Forward | Reverse | |||
| D9Mit121 | GAGCCATGACTCTTGTTTGTAGC | ACAGAGATGGCAGGCTCACT | 127 bp | |
| D9Mit52 | TTTCTGAGTCAGCCAAGGCT | GACTGCTCTTCATGTTTGATGC | 172 bp | |
| D18Mit27 | GTGATTGAATGCTACTAACTCAGCC | CCTCAGGCCTCCACAAATAA | 140 bp | |
| D18Mit15 | CAGACTTCATAGCAACACCCTG | TAACATGAAAACAGAAACAGCCA | 157 bp | |
| D17Mit55 | TCCTGCATTTTTGTGCATGT | CAATGACAAAGAGGAAACAGTCC | 170 bp | |
| D17Mit100 | GTTAAGAATGATTTTCACACTACAAGA | AGCACATGTACTTACTCATATACGTGC | 122 bp | |
| D11Mit161 | TGGAAAAGTGTTACTTTATGGTGTG | AGAGCAACTAAGGAAGGTGCC | 135 bp | |
| Primer name | Sequence | Product size | ||
|---|---|---|---|---|
| Forward | Reverse | |||
| D9Mit121 | GAGCCATGACTCTTGTTTGTAGC | ACAGAGATGGCAGGCTCACT | 127 bp | |
| D9Mit52 | TTTCTGAGTCAGCCAAGGCT | GACTGCTCTTCATGTTTGATGC | 172 bp | |
| D18Mit27 | GTGATTGAATGCTACTAACTCAGCC | CCTCAGGCCTCCACAAATAA | 140 bp | |
| D18Mit15 | CAGACTTCATAGCAACACCCTG | TAACATGAAAACAGAAACAGCCA | 157 bp | |
| D17Mit55 | TCCTGCATTTTTGTGCATGT | CAATGACAAAGAGGAAACAGTCC | 170 bp | |
| D17Mit100 | GTTAAGAATGATTTTCACACTACAAGA | AGCACATGTACTTACTCATATACGTGC | 122 bp | |
| D11Mit161 | TGGAAAAGTGTTACTTTATGGTGTG | AGAGCAACTAAGGAAGGTGCC | 135 bp | |
Sequences of microsatellite primers used in this study
| Primer name | Sequence | Product size | ||
|---|---|---|---|---|
| Forward | Reverse | |||
| D9Mit121 | GAGCCATGACTCTTGTTTGTAGC | ACAGAGATGGCAGGCTCACT | 127 bp | |
| D9Mit52 | TTTCTGAGTCAGCCAAGGCT | GACTGCTCTTCATGTTTGATGC | 172 bp | |
| D18Mit27 | GTGATTGAATGCTACTAACTCAGCC | CCTCAGGCCTCCACAAATAA | 140 bp | |
| D18Mit15 | CAGACTTCATAGCAACACCCTG | TAACATGAAAACAGAAACAGCCA | 157 bp | |
| D17Mit55 | TCCTGCATTTTTGTGCATGT | CAATGACAAAGAGGAAACAGTCC | 170 bp | |
| D17Mit100 | GTTAAGAATGATTTTCACACTACAAGA | AGCACATGTACTTACTCATATACGTGC | 122 bp | |
| D11Mit161 | TGGAAAAGTGTTACTTTATGGTGTG | AGAGCAACTAAGGAAGGTGCC | 135 bp | |
| Primer name | Sequence | Product size | ||
|---|---|---|---|---|
| Forward | Reverse | |||
| D9Mit121 | GAGCCATGACTCTTGTTTGTAGC | ACAGAGATGGCAGGCTCACT | 127 bp | |
| D9Mit52 | TTTCTGAGTCAGCCAAGGCT | GACTGCTCTTCATGTTTGATGC | 172 bp | |
| D18Mit27 | GTGATTGAATGCTACTAACTCAGCC | CCTCAGGCCTCCACAAATAA | 140 bp | |
| D18Mit15 | CAGACTTCATAGCAACACCCTG | TAACATGAAAACAGAAACAGCCA | 157 bp | |
| D17Mit55 | TCCTGCATTTTTGTGCATGT | CAATGACAAAGAGGAAACAGTCC | 170 bp | |
| D17Mit100 | GTTAAGAATGATTTTCACACTACAAGA | AGCACATGTACTTACTCATATACGTGC | 122 bp | |
| D11Mit161 | TGGAAAAGTGTTACTTTATGGTGTG | AGAGCAACTAAGGAAGGTGCC | 135 bp | |
Quantification of mitotic and PCNA indices
Mitotic index was scored on H&E-stained tumors from at least 5 mice/group and at least 2000 hepatocyte nuclei/animal were counted. PCNA-labeling index was determined by counting PCNA-positive cells after counterstaining with hematoxylin and at least 2000 nuclei were counted per mouse. Indices were represented as a percentage of the total cells counted (mean ± SE).
Quantification of tumor size
Tumor size was measured in mm 2 on H&E-stained sections obtained from the central part of hepatocellular carcinoma (HCC) using the ImageJ software.
Quantification of apoptosis
Apoptotic index was calculated by counting the apoptotic figures per 5000 hepatocytes on tumor sections from at least 5 mice/group. Sections were stained with the ApoTag peroxidase in situ apoptosis detection kit (Serologicals, Norcross, GA), and positive cells were counted under light microscope at a magnification 200×. Apoptotic cells, single and closely clustered apoptotic bodies were all scored as single events. Indices were represented as a percentage of the total cells counted (mean ± SE).
Statistical analysis
Student's t test was applied to evaluate statistical significance. Values of P < 0.05 were considered to be statistically significant.
Results
β-Catenin mutations
The frequency of β-catenin gene mutations was determined by PCR amplification and direct sequencing of genomic DNA in 56 HCCs. It was very low in untreated c-myc /TGF-α mice (2/28; 7.1%), but remarkably increased following the PB treatment (9/28; 32.1%). Codon 45 harbored the majority of mutations (81.2 %), which led to a single base transition TCC-TTC (Ser 45→Phe). This mutation has been observed in human HCC, melanoma, thyroid anaplastic carcinoma, desmoid tumors, colorectal cancer and in mouse hepatoblastoma ( 13 ). The other two mutations detected in c-myc /TGF-α-treated HCCs affected the codon 33, resulting in TCT to TAT (Ser 33→Tyr) transversion. There were no interstitial deletions in β-catenin exon 2 in this panel of samples.
Immunohistochemistry and LOH analysis
Since signal transduction via β-catenin involves its translocation into the nucleus ( 13 , 14 ), we next performed immunohistochemistry and determined the sub-cellular localization of β-catenin protein in the same hepatic lesions. Among the panel of examined untreated c-myc /TGF-α HCCs, only tumors with mutated β-catenin (2/28) showed nuclear redistribution of β-catenin consistent with our previous study ( 16 ). The majority of HCCs (18/28; 64.3%) displayed a reduction in β-catenin expression when compared with a strong membranous localization of β-catenin protein in peritumorous dysplastic hepatocytes ( Figure 1A ). To investigate the molecular mechanisms involved in β-catenin down-regulation in c-myc /TGF-α untreated mice, we next determined the microsatellite status of the β-catenin locus in the same collection of HCC samples. Notably, 16/28 (57.1%) HCCs from this group exhibited LOH at the β-catenin locus and all of them showed reduced or absent β-catenin immunostaining. Conversely, PB-treated HCCs did not display any genomic alterations at the same locus ( Figure 2 ).
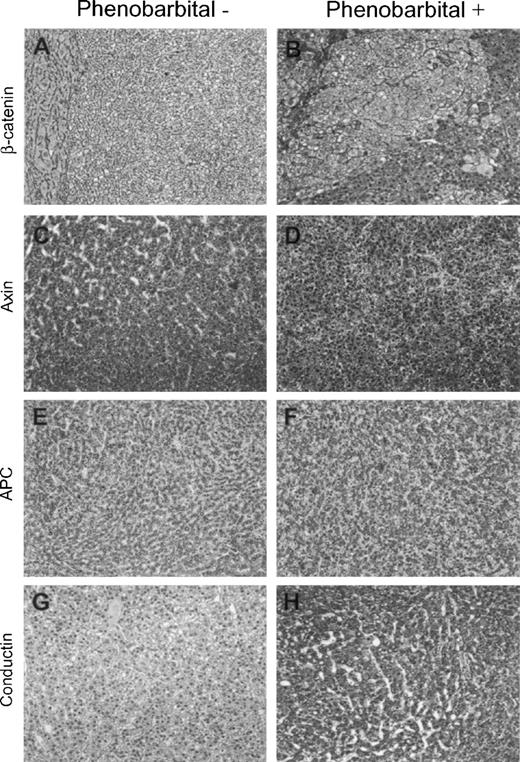
Immunohistochemistry of β-catenin and β-catenin interacting proteins in HCCs from control (Phenobarbital−) or PB-promoted (Phenobarbital+) c-myc /TGF-α mice. ( A ) HCC displaying a marked reduction of β-catenin staining as compared with the surrounding parenchyma (left part of the picture). Magnification 200×. ( B ) HCC from PB-treated mouse showing a strong nuclear and cytoplasmic expression of β-catenin protein. Magnification 250×. No differences were detected in axin ( C and D ) and APC ( E and F ) immunolabeling between untreated and PB promoted HCCs. ( G ) Absence of conductin staining in HCC from a control mouse. ( H ) Strong immunoreactivity for conductin in HCC following PB treatment. Magnification 200× in (C–H). Sections were counterstained with Gill's hematoxylin.
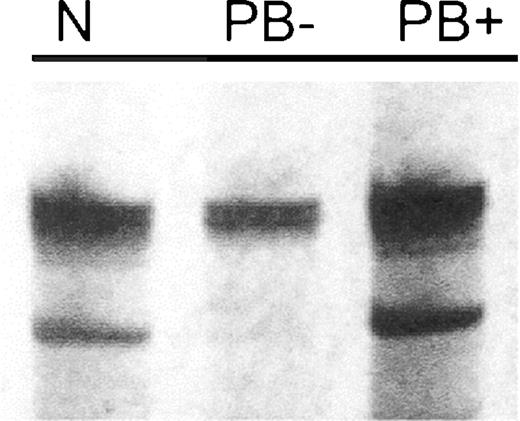
Representative microsatellite analysis of the β-catenin locus in c-myc /TGF-α HCCs. LOH at β-catenin locus in c-myc /TGF-α-untreated (PB−) HCC detected by D9Mit52 microsatellite primers. N, wild-type liver; PB+, HCC promoted by PB.
The PB-treated group of mice exhibited an inappropriate sub-cellular redistribution of β-catenin, which was detected in both β-catenin-mutated tumors and in those lacking exon 2 mutations (15/28; 53.6%). The nuclear localization of wild-type β-catenin suggests that other molecular mechanisms might be involved in its transcriptional activation ( Figure 1B ). Therefore, we examined whether the changes in expression of β-catenin degradation complex members ( 14 ) may account for nuclear accumulation of wild-type β-catenin. There were no major differences in the pattern of APC and axin immunoreactivity in both PB-treated and -untreated tumors. In all examined cases, HCCs displayed cytoplasmic APC and axin immunolabeling ( Figure 1C–F ). Accordingly, no LOH at APC, axin and conductin loci was detected. However, all HCCs with β-catenin nuclear positivity displayed a marked up-regulation of conductin ( Figure 1G and H ). This finding is in agreement with axin2 (human homologous of mouse conductin) over-expression in colon and liver tumors harboring β-catenin defects ( 20 – 22 ). Conductin induction acts as a feedback inhibitor of the Wnt signaling pathway, but its negative regulatory role is either abolished or annulled by alternative overriding pathways ( 21 , 22 ). Moreover, it has been suggested that conductin up-regulation might have paradoxical growth promoting effects on cancer cells ( 22 ).
Strikingly, we detected two distinct patterns of localization of cells with β-catenin nuclear positivity: (i) diffuse or scattered presence within tumor (NAd/s) and (ii) selective accumulation at the invasion front (NAinv) found in the majority of HCCs promoted by PB (11/15, 73.3%). NAinv cells showed high nuclear/cytoplasmic ratio and were arranged as septa-like sheets of small cells that subdivided tumor into the areas with either β-catenin nuclear or membranous localization. In particular, NAinv cells infiltrated the surrounding parenchyma ( Figure 3A and B ). In order to determine whether the NAinv neoplastic hepatocytes display features of immature hepatic cells, we stained the same samples with the A6 monoclonal antibody specific for mouse biliary epithelial and oval cells ( 19 ). No A6 immunostaining was detected in the NAinv tumor cells. Only biliary epithelial cells and rare tumor cells with membranous localization of β-catenin were positive for the A6 antibody ( Figure 3C ).
![Immunohistochemistry of c-myc /TGF-α HCCs promoted by PB with β-catenin NAinv pattern. ( A ) Solid HCC displaying tumor invasion front (indicated by arrows). Magnification 40×. ( B ) β-Catenin neoplastic cells are characterized by a high nucleocytoplasmic ratio and smaller size as compared with adjacent tumor hepatocytes. Magnification 400×. ( C ) NAinv hepatocytes are negative for A6 immunostaining. Biliary epithelial cells (inset) and rare tumor cells (indicated by arrows) display A6 immunolabeling. Magnification 600×. ( D ) E-Cadherin cytoplasmic redistribution that co-localize with β-catenin NAinv positive hepatocytes [compare with β-catenin staining in (A)]. Magnification 40×. (Inset) E-Cadherin cytoplasmic immunolabeling is limited to the small cells at the invasion front, whereas the surrounding tumor cells retain E-cadherin membranous localization. Magnification 400×. ( E and F ) HCC displaying membranous β-catenin and E-cadherin immunoreactivity, respectively. Magnification 200×. ( G and H ) Co-localization of MMP-7, MUC1 and β-catenin (inset) immunostaining. Magnification 200×. Histochemical staining was performed on serial sections in (A) and (D), (E) and (F), (G), (H) and inset. Sections were counterstained with Gill's hematoxylin.](https://oup.silverchair-cdn.com/oup/backfile/Content_public/Journal/carcin/25/6/10.1093_carcin_bgh083/1/m_bgh083f3.jpeg?Expires=1716530319&Signature=R3cnwk2pIjba9i2uZdMRZ-sD-BE45yN4SBYBLhTMEAEtYWPvwvl6aG6RdXnCChxyG9uGrOfN0fg260K3PM00j3-Jtcg-SPb-tie4xN-doFuiZZxTXcw6qtOwyXhUAH4er3TaEq-i8pjtS4O42IY8Agnlk6JXBkZ47xGW7okkrM1hCY2vkg-RRvleGhkNOUqRjungNIpj759dIUuxItPjK6SbP1api-zh3dKHyFEuICNYfEyRg~hMKZ-47Cm0YxVdE7YQnXn02GMCCT6pXxwJ6EyrQ8Ix5do52qyQs6ltHaSUBs2n3lHsox~D9X3eDkDRFpnnA3jPuy9PSenI2vfy9Q__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Immunohistochemistry of c-myc /TGF-α HCCs promoted by PB with β-catenin NAinv pattern. ( A ) Solid HCC displaying tumor invasion front (indicated by arrows). Magnification 40×. ( B ) β-Catenin neoplastic cells are characterized by a high nucleocytoplasmic ratio and smaller size as compared with adjacent tumor hepatocytes. Magnification 400×. ( C ) NAinv hepatocytes are negative for A6 immunostaining. Biliary epithelial cells (inset) and rare tumor cells (indicated by arrows) display A6 immunolabeling. Magnification 600×. ( D ) E-Cadherin cytoplasmic redistribution that co-localize with β-catenin NAinv positive hepatocytes [compare with β-catenin staining in (A)]. Magnification 40×. (Inset) E-Cadherin cytoplasmic immunolabeling is limited to the small cells at the invasion front, whereas the surrounding tumor cells retain E-cadherin membranous localization. Magnification 400×. ( E and F ) HCC displaying membranous β-catenin and E-cadherin immunoreactivity, respectively. Magnification 200×. ( G and H ) Co-localization of MMP-7, MUC1 and β-catenin (inset) immunostaining. Magnification 200×. Histochemical staining was performed on serial sections in (A) and (D), (E) and (F), (G), (H) and inset. Sections were counterstained with Gill's hematoxylin.
Given that defects in the cadherin/catenin complex are involved in the acquisition of the invasive phenotype ( 23 ) and that cytoplasmic redistribution of E-cadherin is a marker of cadherin/catenin complex disruption ( 24 ), we have determined E-cadherin localization by immunohistochemical analysis. Importantly, only hepatocytes displaying the NAinv staining pattern showed intense immunoreactivity for E-cadherin in the cytoplasm ( Figure 3D ), whereas all other tumor cells exhibited E-cadherin membranous localization ( Figure 3F ). This observation suggests that the catenin/ cadherin complex is disrupted in NAinv-positive tumor cells. Since the binding of E-cadherin with β-catenin can be decreased either by matrilysin/matrix metalloproteinase-7 (MMP-7)-mediated cleavage ( 25 ) or by MUC1 over-expression ( 26 ), we have investigated whether MMP-7 or MUC1 levels are associated with catenin/cadherin complex disruption. Notably, strong cytoplasmic immunostaining for both MMP-7 and MUC1 was detected only in HCCs displaying the NAinv pattern and co-localized with β-catenin nuclear immunoreactivity ( Figure 3G and H ), whereas NAinv-negative lesions did not display MMP-7 and MUC1 immunolabeling. Taken together, the data suggest a role for β-catenin in intrahepatic invasion of c-myc /TGF-α HCCs promoted by PB.
Cell proliferation, tumor size and apoptosis
A body of evidence suggests that activation of β-catenin signaling is involved in both cell proliferation and inhibition of apoptosis ( 27 , 28 ). To better define the functional role of β-catenin activation during promotion with PB, we determined the mitotic and apoptotic indices in HCCs from the PB-treated group. The HCCs were divided into three groups: (i) HCCs without β-catenin activation, (ii) HCCs with β-catenin mutations and (iii) HCCs with wild-type β-catenin nuclear accumulation. PCNA labeling indices were significantly higher in both β-catenin mutation and β-catenin activation groups when compared with β-catenin wild-type HCCs ( P = 0.0002 and P = 0.0005, respectively; Figure 4A ). Accordingly, both groups showing β-catenin nuclear accumulation displayed statistically higher mitotic indices than the β-catenin negative group ( P = 0.0003 and P = 0.0014, respectively; Figure 4B ). No differences were detected in the proliferation rate between the two groups displaying nuclear translocation of β-catenin.
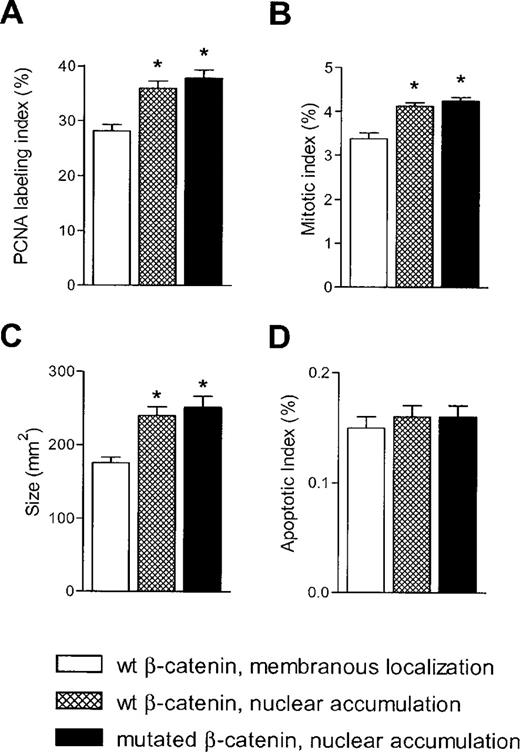
Effect of β-catenin activation on tumor growth in c-myc /TGF-α HCCs promoted by PB. ( A ) PCNA labeling index; ( B ) mitotic index; ( C ) tumor size; ( D ) apoptotic index. Each bar represents mean ± SE ( n = 5–7). *P < 0.002 when compared with HCCs with wild-type β-catenin.
Furthermore, HCCs with deregulated β-catenin exhibited a significantly larger tumor size compared with β-catenin wild-type HCCs ( P = 0.0003; Figure 4C ). However, the apoptotic indices were similar among all three groups of HCCs ( Figure 4D ). These findings indicate that nuclear translocation of β-catenin is associated with an increase in cell proliferation but not with inhibition of apoptosis during c-myc /TGF-α liver carcinogenesis promoted by PB.
Activation of β-catenin in c-myc transgenic mice
We also examined whether the duration of PB treatment may affect the selection for β-catenin defects. For this purpose, we have determined the mutation frequency and cellular localization of β-catenin in HCCs developed in c-myc mice after a short-term promotion with PB. c-myc transgenic mice were fed a NIH-31 diet containing 0.05% PB for 3 weeks starting from 40 days of age. Only two of 20 examined HCCs displayed β-catenin mutations leading to TCC→TTC transition at the codon 45 and an additional three HCCs exhibited nuclear accumulation of β-catenin. Both frequencies, β-catenin mutation (10%) and activation (25%), were very similar to those found previously in HCCs arising in untreated c-myc mice ( 16 ) (11.7 and 23.5%, respectively). The data suggest that only prolonged PB treatment selects for the outgrowth of β-catenin positive neoplastic clones.
Discussion
Our data show that PB treatment increases the frequency of β-catenin mutations in the c-myc /TGF-α model of liver cancer. The prevalence of β-catenin mutations detected in PB-promoted c-myc /TGF-α HCCs (32%) is lower than that described in a mouse model of chemically induced liver cancer (80%) ( 10 ). The reason for the difference in the β-catenin mutation frequency between these two mouse models of HCC is unclear. It is possible that β-catenin mutation incidence depends on the genetic background. However, it seems more likely that deregulation of the β-catenin gene is a fundamental step during DEN + PB-induced hepatocarcinogenesis, whereas several other genetic pathways support the development of liver cancer in c-myc /TGF-α transgenic mice treated with PB. Additionally, promotion with PB leads to activation of the Wnt/β-catenin pathway in c-myc /TGF-α HCCs via molecular mechanisms different from β-catenin mutations. However, no alterations were observed in the members of the β-catenin degradation complex implying that other members of the Wnt signaling pathway may be responsible for the aberrant nuclear accumulation of β-catenin. To the best of our knowledge, this is the first report showing that PB promotes nuclear stabilization of β-catenin protein in the presence of the wild-type β-catenin gene. Taken together, these findings suggest that PB, which is per se non-genotoxic, favors the positive selection of pre-existing neoplastic sub-populations with multiple abnormalities in the Wnt pathway. Furthermore, it seems that only chronic PB treatment is able to select for the β-catenin phenotype, as short-term PB administration did not increase the rate of β-catenin activation in c-myc HCCs.
In HCCs derived from c-myc /TGF-α-untreated mice, activation of β-catenin was a very rare event. Moreover, the expression of β-catenin in tumor tissue was often lower than in the surrounding liver, in agreement with our previous report ( 16 ). The majority of HCCs with lost or reduced immunolabeling of β-catenin (16/18, 89%) exhibited LOH at the β-catenin locus suggesting that these two events are closely related.
Significantly, we found the presence of two distinct localization patterns of cells with β-catenin nuclear accumulation in PB-treated c-myc /TGF-α HCCs. The first, characterized by the presence of scattered or numerous malignant hepatocytes with nuclear translocation of β-catenin, has been described in HCCs from c-myc , c-myc /TGF-α and c-myc /TGF-β1 transgenic mice ( 14 ) whereas the second has been reported previously only for human colon cancer ( 29 ). The prominent feature of the second staining pattern was the presence of sheets composed of small hepatocytes with β-catenin nuclear localization, which were found at the tumor invasion front. Importantly, NAinv positive hepatocytes displayed E-cadherin cytoplasmic redistribution associated with MUC1 and MMP-7 over-expression. MUC1, an integral membrane mucin associated with the metastatic phenotype, is known to interact with β-catenin and promote cellular invasion ( 30 ). In vitro studies have shown that activation of β-catenin up-regulates transcription of MMP-7, a member of the matrix metalloproteinase family, that directly participates in the process of invasion and metastasis ( 15 ). Furthermore, recent studies have demonstrated that metalloproteinase 1 and 26 are β-catenin-specific targets ( 31 , 32 ). Taken together, the data suggest that β-catenin may play a novel role underlying the transformation and invasion in c-myc /TGF-α liver lesions promoted by PB, supporting findings in human colon and breast carcinoma ( 29 , 30 ). β-Catenin/MUC1 interaction may result in the disruption of cell adhesion, promoting neoplastic invasion and allowing for tumor expansion in c-myc /TGF-α HCCs. MMP-7 induction may further facilitate this process through its ability to dissociate the catenin/cadherin complex ( 25 ). However, other molecular mechanisms might contribute to the disruption of the catenin/cadherin binding, including increased activity of src ( 33 ) or over-expression of HGF ( 34 ).
The role of the Wnt pathway in the liver is still poorly understood, as neither c-myc , nor cyclin D1, well-known β-catenin target genes, are induced in β-catenin benign and malignant hepatic lesions ( 10 , 35 ). Current evidence indicates that β-catenin activation can control both hepatocyte growth and survival ( 36 , 37 ). In particular, it has been suggested from the correlative study linking β-catenin activation and up-regulation of glutamine synthetase that the β-catenin phenotype confers selective growth advantages during liver tumor development ( 38 , 39 ). In c-myc /TGF-α HCCs, we have shown that β-catenin activation was associated with increased cell proliferation and tumor size. The data suggest that activation of Wnt signaling provides proliferative rather than anti-apoptotic advantages in β-catenin-positive c-myc /TGF-α tumors. β-Catenin transactivation may also correlate with resistance to apoptosis as has been found in HCCs from c-myc /E2F-1 transgenic mice (D.F.Calvisi et al ., in preparation). Therefore, it appears that β-catenin aberrant activation exerts either a proliferative or an anti-apoptotic function, depending upon the tumor model and its biology. Thus, activation of the NF-κB/Akt pathway leads to a strong inhibition of apoptosis during c-myc /TGF-α tumor development ( 40 ), rendering the survival role of β-catenin superfluous. In contrast, β-catenin provides a sustained anti-apoptotic stimulus in c-myc /E2F-1-driven hepatocarcinogenesis where the NF-κB pathway is not active (D.F.Calvisi et al ., in preparation).
In summary, the data show that activation of β-catenin confers proliferative and invasive advantages during liver tumor promotion by PB in c-myc /TGF-α transgenic mice and suggest a mechanism by which PB can accelerate hepatic tumor evolution and progression.
D.F.C. was supported in part by a fellowship from FIRC (Fondazione Italiana per la Ricerca sul Cancro).
References
Jirtle,R.L., Meyer,S.A. and Brockenbrough,J.S. (
Brockenbrough,J.S., Meyer,S.A., Li,C.X. and Jirtle,R.L. (
Bursch,W., Lauer,B., Timmermann-Trosiener,I., Barthel,G., Schuppler,J. and Schulte-Hermann,R. (
Guppy,M.J., Wilton,J.C., Sharma,R., Coleman,R. and Chipman,J.K. (
Murakami,H., Sanderson,N.D., Nagy,P., Marino,P.A., Merlino,G. and Thorgeirsson,S.S. (
Santoni-Rugiu,E., Nagy,P., Jensen,M.R., Factor,V.M. and Thorgeirsson,S.S. (
Santoni-Rugiu,E., Jensen,M.R. and Thorgeirsson,S.S. (
Sanders,S. and Thorgeirsson,S.S. (
Sanders,S. and Thorgeirsson,S.S. (
Aydinlik,H., Nguyen,T.D., Moennikes,O., Buchmann,A. and Schwarz,M. (
Ozawa,M. and Kemler,R. (
Chen,H., Paradies,N.E., Fedor-Chaiken,M. and Brackenbury,R. (
Kikuchi,A. (
Crawford,H.C., Fingleton,B.M., Rudolph-Owen,L.A., Goss,K.J., Rubinfeld,B., Polakis,P. and Matrisian,L.M. (
Calvisi,D.F., Factor,V.M., Loi,R. and Thorgeirsson,S.S. (
Frith,C.H., Ward,J.M. and Turusov,V.S. (
De La Coste,A., Romagnolo,B., Billuart,P. et al . (
Engelhardt,N.V., Factor,V.M., Yasova,A.K., Poltoranina,V.S., Baranov,V.N. and Lasareva,M.N. (
Yan,D., Wiesmann,M., Rohan,M. et al . (
Lustig,B., Jerchow,B., Sachs,M. et al . (
Leung,J.Y., Kolligs,F.T., Wu,R., Zhai,Y., Kuick,R., Hanash,S., Cho,K.R. and Fearon,E.R. (
Behrens,J. (
Nawrocki,B., Polette,M., Van Hengel,J., Tournier,J.M., Van Roy,F. and Birembault,P. (
Davies,G., Jiang,W.G. and Mason,M.D. (
Li,Y., Bharti,A., Chen,D., Gong,J. and Kufe,D. (
Miller,J.R., Hocking,A.M., Brown,J.D. and Moon,R.T. (
Giles,R.H., van Es,J.H. and Clevers,H. (
Ougolkov,A.V., Yamashita,K., Mai,M. and Minamoto,T. (
Schroeder,J.A., Adriance,M.C., Thompson,M.C., Camenisch,T.D. and Gendler,S.J. (
Takahashi,M., Tsunoda,T., Seiki,M., Nakamura,Y. and Furukawa,Y. (
Marchenko,G.N., Marchenko,N.D., Leng,J. and Strongin,A.Y. (
Irby,R.B. and Yeatman,TJ. (
Hiscox,S. and Jiang,W.G. (
Cadoret,A., Ovejero,C., Saadi-Kheddouci,S., Souil,E., Fabre,M., Romagnolo,B., Kahn,A. and Perret,C. (
Monga,S.P., Pediaditakis,P., Mule,K., Stolz,D.B. and Michalopoulos,G.K. (
Monga,S.P., Monga,H.K., Tan,X., Mule,K., Pediaditakis,P. and Michalopoulos,G.K. (
Cadoret,A., Ovejero,C., Terris,B., Souil,E., Levy,L., Lamers,W.H., Kitajewski,J., Kahn,A. and Perret,C. (
Loeppen,S., Schneider,D., Gaunitz,F., Gebhardt,R., Kurek,R., Buchmann,A. and Schwarz,M. (

{kind=link}
{kind=link}
{kind=link}
{kind=link}