




Abstract
Short-term dextran sodium sulfate (DSS) treatment has been shown to notably accelerate colorectal tumor development in rats initiated with 1,2-dimethylhydrazine (DMH). In the present study, to clarify mechanisms underlying the DSS influence, time-course studies of histopathological and immunohistochemical characteristics and β-catenin gene mutations in colorectal mucosa in early stages of this model were conducted. F344 males were given three subcutaneous injections of DMH (40 mg/kg body wt) within a week, followed by free access to drinking water containing 1% DSS for a week. At weeks 1, 4, 6 and 8 after the DSS treatment, rats were euthanized and colorectal samples were collected. At week 1, the colorectal mucosa demonstrated extensive erosion along with significant inflammatory cell infiltration and neighboring reactive hyperplasia. By week 4, the mucosal damage was repaired and regenerative mucosa, partly characterized by Paneth cell metaplasia and altered subcellular localization of β-catenin, was apparent. Areas with Paneth cells/β-catenin accumulation were significantly more likely to be accompanied by interstitial inflammation and 17 of 24 dysplastic foci were found in regenerative mucosa with Paneth cells. Furthermore, adenomas/carcinomas frequently featured various degrees of Paneth cell differentiation. Point mutations mainly in codons 34 and 41 of β-catenin gene were detected in 6 of 27 samples of regenerative mucosa with Paneth cells and four of nine dysplastic foci/adenomas/carcinomas. These findings indicate that inflammation-associated regenerative mucosa with Paneth cell metaplasia and alteration in the APC/β-catenin/Tcf signal transduction pathway are possibly involved in the acceleration of colorectal carcinogenesis in this DMH–DSS rat model.
Introduction
Patients with chronic inflammatory bowel disease (IBD), which includes Crohn's disease and ulcerative colitis (UC), are at increased risk of developing colorectal cancer ( 1 , 2 ). Most IBD-associated cancers tend to arise within mucosa affected by colonic inflammation ( 2 , 3 ), with precursors recognized as low- to high-grade dysplasias that may spread over large areas ( 2 , 4 ). Although morphological characteristics during remodeling of regenerated colorectal mucosa of IBD patients have been well documented ( 5–7 ), molecular events in early processes of carcinogenesis before development of dysplasia have yet to be clearly defined and how chronic inflammation actually contributes to tumor development remains to be clarified ( 8 ).
Somatic mutations of tumor suppressor adenomatous polyposis coli ( APC ) genes, which are known to play crucial roles in sporadic colorectal carcinogenesis ( 9 , 10 ), and APC-associated β-catenin mutations, frequently detected in colorectal tumors lacking APC mutations ( 11 , 12 ), have been demonstrated to be rare in UC-related tumors ( 13 ). Hypermethylation of the APC gene has also been shown in sporadic colorectal carcinomas ( 14 ), but this has not yet been demonstrated in Crohn's disease or UC-related lesions. On the other hand, assessment of allele loss at microsatellite markers close to known or putative tumor suppressor genes and immunohistochemical analysis in UC-associated dysplasias and carcinomas revealed moderate to high frequencies of allele loss near APC (31%, n = 48) or β-catenin (27%, n = 55) genes and altered expression of APC and β-catenin proteins in both UC-related and sporadic tumors ( 15 , 16 ), suggesting that some alterations in the genes associated with the APC/β-catenin/Tcf signal transduction pathway might occur in UC-related carcinogenesis.
To establish experimental models relevant to colitis-associated neoplasia in humans, oral administration of dextran sodium sulfate (DSS), a synthetic sulfated polysaccharide composed of dextran with sulfated anhydroglucose units, to mice, rats or hamsters, with or without pretreatment of colon carcinogens, has been widely utilized ( 17–23 ). Since DSS is assumed to be non-genotoxic ( 24 , 25 ) but reported to induce erosion, ulceration and/or inflammation of colorectal mucosa in mice and rats ( 19 , 25 , 26 ), its effects are at least partly due to tumor promotion as a result of regenerative proliferation. However, a 7 day DSS treatment was shown to be sufficient to induce dysplasias after a 180 day withdrawal period in mice ( 27 ), so that irreversible molecular events may also occur in association with DSS-induced mucosal damage and/or inflammation. Information concerned with mechanisms underlying DSS-induced colorectal carcinogenesis has recently accumulated from studies of mouse models ( 28–30 ).
In rat, colon carcinogenesis induced by genotoxic carcinogens, several morphological and molecular events in early stages have also been well documented. As putative pre-neoplastic lesions, aberrant crypt foci ( 31 ), β-catenin accumulated crypts ( 32 ), mucin-depleted foci ( 33 ), dysplastic aberrant crypt foci ( 34 ) and flat dysplastic aberrant crypt foci ( 35 ) have been proposed. As a genetic alteration, mutation of β-catenin in such pre-neoplastic lesions is reported to be an early event ( 34 , 36–38 ). On the other hand, morphological and molecular investigations of colitis-induced rat models have been limited. Recently, we have established a rapid carcinogenesis model in rats initiated with 1,2-dimethylhydrazine (DMH) followed by a 1 week DSS treatment, in which neoplastic lesions including adenocarcinomas localized in distal parts of the colon/rectum can be induced within 10 weeks ( 39 ). This model has been utilized for detection of colorectal carcinogenesis modifiers, applying neoplastic lesions as end-points, within a short-term period ( 40 ), but possible mechanisms of the DSS-induced colitis-associated tumor promotion have yet to be explored in detail. With respect to roles of the APC/β-catenin pathway in DSS-associated carcinogenesis, immunohistochemistry for β-catenin revealed an altered subcellular localization in neoplasms in some mouse models treated with DSS with or without carcinogen pretreatment ( 28 , 29 ), but early events in the colorectal mucosa are not completely understood. In the present study, to characterize the early morphological characteristics and genetic events of colorectal mucosa in the DMH–DSS rat model, time-course histopathological analyses, with immunohistochemistry for β-catenin and genetic analysis for β-catenin , were performed. In addition, cell proliferation kinetics in the reactive hyperplastic/regenerative mucosa after DSS-induced damage were analyzed with reference to expression of β-catenin, known to regulate epithelial cell growth ( 41 , 42 ).
Materials and methods
Chemicals and animals
DMH was purchased from Tokyo Kasei Kogyo (Tokyo, Japan) and DSS (Molecular weight 36 000–50 000) was from ICN Biochemicals (Aurora, OH). A total of 28 male F344 rats at 6 weeks of age were purchased from Japan SLC (Shizuoka, Japan) and housed four rats per polycarbonate cage with white wood chips (Sankyo Laboratory Service, Tokyo, Japan) for bedding in a standard air-conditioned room (24 ± 1°C, 55 ± 5% relative humidity, 12 h light and dark cycle), with free access to basal diet (CRF-1; Oriental Yeast, Tokyo, Japan) and tap water.
Experimental protocol
Twenty of 28 animals were given three subcutaneous injections of DMH (40 mg/kg body wt, dissolved at 8 mg/ml in saline) in the first week and allowed free access to deionized water containing 1% DSS for 1 week thereafter, as described earlier ( 40 ). These animals were then given basal diet and tap water ad libitum . At weeks 1, 4, 6 and 8 after DSS treatment, 4, 8, 4 and 4 rats, respectively, were euthanized by exsanguination under deep ether anesthesia for collection of colorectal samples. The remaining eight non-initiated animals were maintained without DMH or DSS treatment and four animals each were euthanized at weeks 1 and 4 for control sample collection. At autopsy, the entire colon and rectum of each animal were excised, opened longitudinally, stretched flat with needles on styrofoam board and fixed in 10% neutral buffered formalin.
Histopathological observations
The fixed entire colons and rectums of four DMH–DSS-treated animals at each time point and control animals at week 1 were cut into three equal lengths from the proximal to the distal ends, and the distal part was additionally cut longitudinally into two strips and routinely embedded in paraffin. Serial sections were prepared for staining with hematoxylin and eosin, immunohistochemistry and/or DNA extraction. The middle and proximal colon were cut longitudinally into two strips and similarly processed to hematoxylin and eosin stained sections. Proportions of regenerative mucosa with or without Paneth cell metaplasia in total mucosal (muscularis musosae) length of the sectioned distal colon and rectum of each animal and proportions of regions accompanied by interstitial inflammation in the regenerative mucosa with or without Paneth cell metaplasia were measured with an IPAP-WIN morphometric analyzer (Sumika Technoservice, Hyogo, Japan). The fixed distal part of the remaining four DMH–DSS-treated and control animals each at week 4 was processed into serial paraffin sections of the middle to lower crypt zone of mucosa by en face preparation ( 36 ) and stained with hematoxylin and eosin and immunohistochemistry, to investigate architectural characteristics of regenerative mucosa and location of pre-neoplastic lesions in large mucosal areas. Pre-neoplastic and neoplastic lesions of colorectal mucosa were histopathologically classified into dysplastic foci, adenomas and adenocarcinomas as described earlier in rat colorectal carcinogenesis models ( 20 , 25 , 34 ).
Immunohistochemistry
Monoclonal antibody against β-catenin was purchased from BD Transduction Laboratories (clone 14, Lexington, KY) used at a dilution of 1/500 and an anti-rat Ki-67 antigen for determination of cell proliferative activities from DAKO (clone MIB-5, Glostrup, Denmark) used at 1/100. Antigen retrieval was performed in an autoclave for 15 min at 121°C in 10 mM citrate buffer (pH 6.0) for both β-catenin and Ki-67. The streptavidin–biotin peroxidase complex method (StreptABComplex/HRP, DAKO) or a peroxidase-labeled amino acid polymer method (Histofine simple stain rat MAX-PO, Nichirei Bioscience, Tokyo, Japan) was used to determine the expression and localization of each antigen, and sections were lightly counterstained with hematoxylin for microscopic examination. Negative controls were included without primary antibodies for each antigen using serial sections. In the lesions sampled for mutation analysis described below, immunohistochemical intensity for β-catenin was evaluated as weak to moderate, which means ≤50% of crypts contained β-catenin-positive epithelial cells, or strong, which means >50% positive cells in each lesion. Ki-67-positive nuclei per 300–1400 epithelial cells were counted in randomly selected regions of normal-appearing mucosa and reactive hyperplastic mucosa from all DMH–DSS-treated animals at week 1 and of normal-appearing mucosa and regenerative mucosa with and without Paneth cells from DMH–DSS-treated animals at weeks 4 and 6.
Mutation analysis of β-catenin
DNA extraction from paraffin-embedded sections was performed as follows. Lesions were dissected under a microscope using the Pinpoint Slide DNA Isolation System (Zymo Research, Orange, CA) according to the manufacturer's protocol. Extracted DNA was then subjected to polymerase chain reaction (PCR)-based direct sequencing using the CEQ2000XL DNA analysis system (Beckman Coulter, Fullerton, CA). PCR primers for the rat β-catenin gene were designed to amplify the 193 bp fragment from exon 3, corresponding to phosphorylation sites. Primer sequences were 5′-TGACCTCATGGAGTTGGACA-3′ for forward, 5′-GCCTTGCTCCCACTCATAAA-3′ for reverse and with an annealing temperature of 60°C. PCR reactions were performed using AmpliTaq Gold DNA polymerase (Applied Biosystems Japan, Tokyo, Japan). Nested sequence primers used for direct sequencing of the PCR products were 5′-CAGACAGAAAGGCCGCTGT-3′ and 5′-CATCTTCTTCCTCAGGATTGC-3′ for forward and reverse, respectively. Mutation analyses were performed twice for forward and once for reverse to confirm the results.
Statistical analysis
Statistical analysis to compare the proportions of regenerative mucosa with or without Paneth cell metaplasia, the proportions of regions accompanied by interstitial inflammation in the regenerative mucosa with or without Paneth cell metaplasia and the Ki-67 labeling indices was performed with the Student's or Welch's t -test following the F test. Significance was inferred at the 5%, 1% and 0.1% levels.
Results
General conditions and body weight
After 1 week of DSS treatment, a few rats showed bloody/soft stools. Thereafter, no such clinical symptoms were observed. Body weight gain of DMH–DSS-treated animals was significantly ( P < 0.05) smaller than the no-treatment controls (initial body weights and values week 1 after DSS treatment were 135.4 ± 5.5 and 197.4 ± 14.7 g, respectively, for DMH–DSS-treated rats and 135.6 ± 4.1 and 218.2 ± 5.6 g for control rats).
Gross findings and histopathology
At necropsy, after 1 week of DSS treatment, no obvious macroscopic changes were detected, except for slight roughening of the surface in the distal parts of the colon and rectal mucosa of DMH–DSS-treated animals. At weeks 6 and 8, polypoid/protruded nodules were observed in the distal part of the colon and rectal mucosa of 2 of 4 animals each. In a total of three animals without nodules, a cloudy area of mucosal surface continuous from anus to rectum/distal colon, corresponding to histopathological squamous metaplasia described below, was visible at weeks 4 and 6. Also, on microscopic observation, major changes were observed in the distal colon and rectum. At week 1, regional loss of crypts, so-called erosion, accompanied by severe inflammatory cell infiltration, consisting mainly of neutrophils and foamy mononuclear cells, and various degrees of edema and fibrosis in the lamina propria and/or submucosa, was noted in all DMH–DSS-treated animals ( Figure 1A ). Reactive mucosal hyperplasia accompanied by mucosal thickening in areas neighboring erosion was also frequently detected in all animals. Some crypts in the reactive hyperplastic mucosa were simply elongated and lined by hyperplastic epithelium with goblet cells, while some featured an irregular glandular structure lined by hyperplastic basophilic epithelium with neither goblet cells nor distinct cellular atypia ( Figure 1B ). Others were intermediate in appearance. At weeks 4–8, mucosal damage and pronounced inflammatory cell infiltration were no longer evident. Instead, regenerative mucosa showing abnormal cryptal architecture, with distortion, dilatation, branching and/or shortening of crypts, with or without mucosal atrophy and variation in inter-crypt spacing, was observed ( Figures 2A, D and G and 3A ). The regenerative mucosa was characterized by minimal cellular atypia and goblet cell differentiation. Especially in some regions with interstitial inflammatory cell infiltration, Paneth cell metaplasia characterized by cytoplasmic eosinophilic granules was frequently seen ( Figure 2G and 3A ). Data for proportions of regenerative mucosa with or without Paneth cell metaplasia in longitudinally embedded sections at each time point are summarized in Table I . The values regenerative mucosa with Paneth cells peaked at week 4, with tendencies to decrease thereafter. Regenerative mucosa with Paneth cells was significantly ( P < 0.01) more associated with interstitial inflammation than regenerative mucosa without Paneth cells ( Table I ). The Paneth cells appeared mainly in the lower crypt zone but also in the middle zone in the longitudinally embedded sections ( Figure 2G ), and in the sections by an en face preparation they were frequently observed in laterally branching crypts in the middle zone ( Figure 3B ). Dysplastic foci, accompanied by moderate cellular atypia and almost lacking goblet cells, arose at weeks 4–8 and adenomas and adenocarcinomas at weeks 6 and 8 ( Table II ). Eight of 11 (73%) dysplastic foci found in the routinely longitudinally embedded sections on weeks 4–8 and nine of 13 (69%) dysplastic foci found in the sections by an en face preparation on week 4 were surrounded by regenerative mucosa with Paneth cells ( Figures 3A ). Eleven of 24 (46%) dysplastic foci, three of 4 adenomas and one adenocarcinoma featured various degrees of Paneth cell differentiation. The macroscopic cloudy mucosal surface observed on weeks 4 and 6 was confirmed as squamous metaplasia of the colorectal mucosa, continuous from the anus. Microscopic findings in the middle and proximal colon were limited to diffuse mucosal thickening and increased size of mucosal lymphoid follicles at weeks 1–6 and weeks 1–8, respectively.
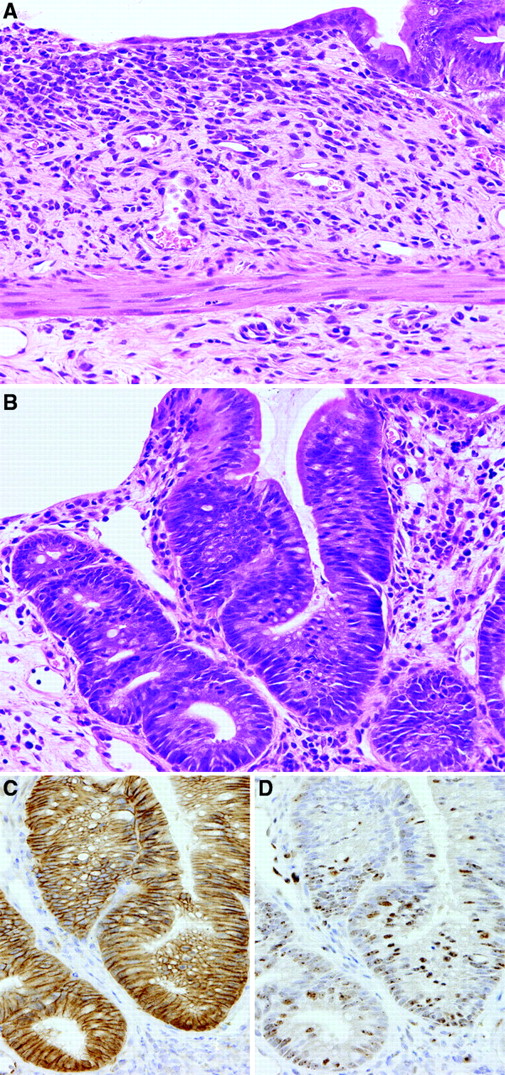
Colorectal mucosal lesions of rats after week 1 of DSS treatment following DMH initiation. ( A ) Regional erosion accompanied by inflammatory cell infiltration, edema and fibrosis in lamina propria and/or submucosa. Hematoxylin and eosin. Original magnification, ×180. ( B ) Reactive mucosal hyperplasia featuring an irregular glandular structure without distinct cellular atypia. Hematoxylin and eosin. Original magnification, ×180. ( C ) A serial section of ( B ). Note immunoreactivity of β-catenin in the epithelial cells. β-catenin—immunohistochemistry. ( D ) A serial section of ( B ). Distinct Ki-67 positivity is shown in the reactive hyperplastic epithelial cells. Ki-67—immunohistochemistry.
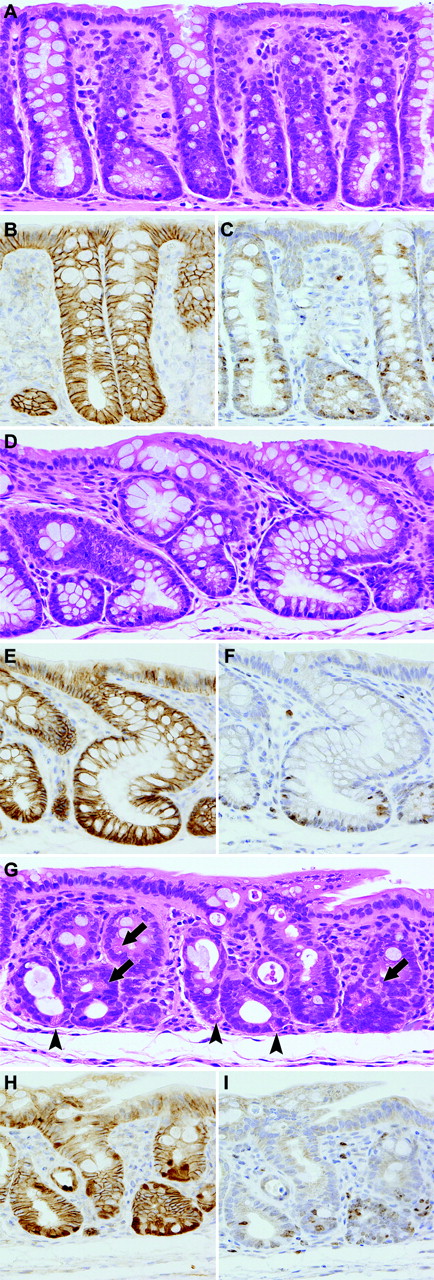
Colorectal mucosal lesions at week 4–6 in routinely longitudinally embedded sections after DSS treatment following DMH initiation. ( A ) Normal mucosa of a non-treated control rat. Hematoxylin and eosin. Original magnification, ×180. ( B ) A serial section of ( A ). Note immunoreactivity of β-catenin in the epithelial cells. β-catenin—immunohistochemistry. ( C ) A serial section of ( A ). Note Ki-67-positive cells in the lower crypt zone. Ki-67—immunohistochemistry. ( D ) Regenerative mucosa showing distortion and/or dilatation of crypts without distinct cellular atypia. Hematoxylin and eosin. Original magnification, ×180. ( E ) A serial section of ( D ). Note immunoreactivity of β-catenin in the epithelial cells. β-catenin—immunohistochemistry. ( F ) A serial section of ( D ). Note Ki-67-positive cells in the lower crypt zone. Ki-67—immunohistochemistry. ( G ) Regenerative mucosa with Paneth cell metaplasia, characterized by cytoplasmic eosinophilic granules, in the lower crypt zone (arrowheads) and middle zone (arrows). Crypts show distortion and shortening. Hematoxylin and eosin. Original magnification, ×180. ( H ) A serial section of ( G ). Nuclear/cytoplasmic expression of β-catenin in subsets of epithelial cells. β-catenin—immunohistochemistry. ( I ) A serial section of ( G ). Note Ki-67-positive cells in the lower crypt zone and the middle zone. Ki-67—immunohistochemistry.
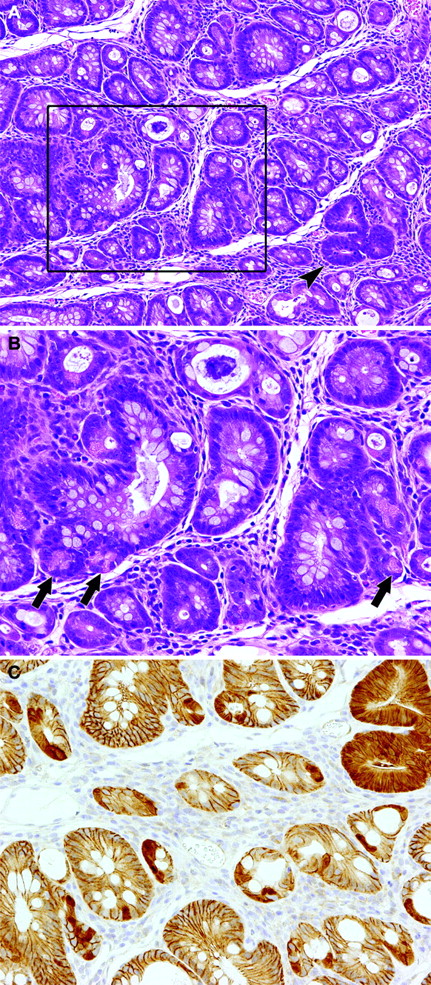
Colorectal mucosal lesions in sections after en face preparation at week 4 after DSS treatment following DMH initiation. ( A ) Regenerative mucosa with interstitial inflammation and Paneth cell metaplasia. A dysplastic focus (arrowhead) is also included. Hematoxylin and eosin, ×90. ( B ) A higher magnification of the box in ( A ). Paneth cells frequently observed in laterally branching crypts (arrows), ×180. ( C ) A serial section of ( A ). Note nuclear/cytoplasmic expression of β-catenin in the dysplastic focus and subsets of epithelial cells in the regenerative mucosa with Paneth cells. β-catenin—immunohistochemistry, ×180.
Proportions of regenerative mucosa with or without Paneth cell metaplasia in the distal colon and rectal mucosa of rats treated with DSS after DMH initiation
| Week after DSS administration | No. of rats analyzed | Regenerative mucosa | |||||
| With Paneth cell metaplasia | Without Paneth cell metaplasia | ||||||
| Incidence | % of total mucosal length | % of length accompanied by interstital inflammation | Incidence | % of total mucosal length | % of length accompanied by interstital inflammation | ||
| 1 | 4 | 0 | — | — | 0 | — | — |
| 4 | 4 | 4 | 17.9 ± 8.1 | 42.2 ± 14.1 a | 4 | 32.5 ± 8.4 | 8.7 ± 5.6 |
| 6 | 4 | 4 | 8.8 ± 7.8 | 38.3 ± 26.6 | 4 | 32.4 ± 11.3 | 18.2 ± 21.5 |
| 8 | 4 | 2 | 5.6 ± 10.3 | 13.8 ± 27.6 | 3 | 13.7 ± 10.3 | 4.5 ± 9.0 |
| Control | 4 | — | — | ||||
| Week after DSS administration | No. of rats analyzed | Regenerative mucosa | |||||
| With Paneth cell metaplasia | Without Paneth cell metaplasia | ||||||
| Incidence | % of total mucosal length | % of length accompanied by interstital inflammation | Incidence | % of total mucosal length | % of length accompanied by interstital inflammation | ||
| 1 | 4 | 0 | — | — | 0 | — | — |
| 4 | 4 | 4 | 17.9 ± 8.1 | 42.2 ± 14.1 a | 4 | 32.5 ± 8.4 | 8.7 ± 5.6 |
| 6 | 4 | 4 | 8.8 ± 7.8 | 38.3 ± 26.6 | 4 | 32.4 ± 11.3 | 18.2 ± 21.5 |
| 8 | 4 | 2 | 5.6 ± 10.3 | 13.8 ± 27.6 | 3 | 13.7 ± 10.3 | 4.5 ± 9.0 |
| Control | 4 | — | — | ||||
Mean ± SD.
The values are based on the findings with longitudinally embedded sections.
P < 0.01 versus regenerative mucosa without Paneth cell metaplasia (Student's t -test).
Proportions of regenerative mucosa with or without Paneth cell metaplasia in the distal colon and rectal mucosa of rats treated with DSS after DMH initiation
| Week after DSS administration | No. of rats analyzed | Regenerative mucosa | |||||
| With Paneth cell metaplasia | Without Paneth cell metaplasia | ||||||
| Incidence | % of total mucosal length | % of length accompanied by interstital inflammation | Incidence | % of total mucosal length | % of length accompanied by interstital inflammation | ||
| 1 | 4 | 0 | — | — | 0 | — | — |
| 4 | 4 | 4 | 17.9 ± 8.1 | 42.2 ± 14.1 a | 4 | 32.5 ± 8.4 | 8.7 ± 5.6 |
| 6 | 4 | 4 | 8.8 ± 7.8 | 38.3 ± 26.6 | 4 | 32.4 ± 11.3 | 18.2 ± 21.5 |
| 8 | 4 | 2 | 5.6 ± 10.3 | 13.8 ± 27.6 | 3 | 13.7 ± 10.3 | 4.5 ± 9.0 |
| Control | 4 | — | — | ||||
| Week after DSS administration | No. of rats analyzed | Regenerative mucosa | |||||
| With Paneth cell metaplasia | Without Paneth cell metaplasia | ||||||
| Incidence | % of total mucosal length | % of length accompanied by interstital inflammation | Incidence | % of total mucosal length | % of length accompanied by interstital inflammation | ||
| 1 | 4 | 0 | — | — | 0 | — | — |
| 4 | 4 | 4 | 17.9 ± 8.1 | 42.2 ± 14.1 a | 4 | 32.5 ± 8.4 | 8.7 ± 5.6 |
| 6 | 4 | 4 | 8.8 ± 7.8 | 38.3 ± 26.6 | 4 | 32.4 ± 11.3 | 18.2 ± 21.5 |
| 8 | 4 | 2 | 5.6 ± 10.3 | 13.8 ± 27.6 | 3 | 13.7 ± 10.3 | 4.5 ± 9.0 |
| Control | 4 | — | — | ||||
Mean ± SD.
The values are based on the findings with longitudinally embedded sections.
P < 0.01 versus regenerative mucosa without Paneth cell metaplasia (Student's t -test).
Incidences and numbers of pre-neoplastic and neoplastic lesions in the distal colon and rectal mucosa of rats treated with DSS after DMH initiation
| Week after DSS administration | No. of rats analyzed | Dysplastic foci | Adenoma | Adenocarcinoma | |||
| Incidence | Number/rat | Incidence | Number/rat | Incidence | Number/rat | ||
| 1 | 4 | 0 | — | 0 | — | 0 | — |
| 4 | 4 | 1 | 0.3 ± 0.1 | 0 | — | 0 | — |
| 6 | 4 | 3 | 1.8 ± 1.7 | 1 | 0.3 ± 0.1 | 1 | 0.3 ± 0.1 |
| 8 | 4 | 2 | 0.8 ± 1.0 | 2 | 0.8 ± 1.0 | 0 | — |
| Control | 4 | — | — | — | |||
| Week after DSS administration | No. of rats analyzed | Dysplastic foci | Adenoma | Adenocarcinoma | |||
| Incidence | Number/rat | Incidence | Number/rat | Incidence | Number/rat | ||
| 1 | 4 | 0 | — | 0 | — | 0 | — |
| 4 | 4 | 1 | 0.3 ± 0.1 | 0 | — | 0 | — |
| 6 | 4 | 3 | 1.8 ± 1.7 | 1 | 0.3 ± 0.1 | 1 | 0.3 ± 0.1 |
| 8 | 4 | 2 | 0.8 ± 1.0 | 2 | 0.8 ± 1.0 | 0 | — |
| Control | 4 | — | — | — | |||
Mean ± SD.
The values are based on findings with longitudinally embedded sections.
Incidences and numbers of pre-neoplastic and neoplastic lesions in the distal colon and rectal mucosa of rats treated with DSS after DMH initiation
| Week after DSS administration | No. of rats analyzed | Dysplastic foci | Adenoma | Adenocarcinoma | |||
| Incidence | Number/rat | Incidence | Number/rat | Incidence | Number/rat | ||
| 1 | 4 | 0 | — | 0 | — | 0 | — |
| 4 | 4 | 1 | 0.3 ± 0.1 | 0 | — | 0 | — |
| 6 | 4 | 3 | 1.8 ± 1.7 | 1 | 0.3 ± 0.1 | 1 | 0.3 ± 0.1 |
| 8 | 4 | 2 | 0.8 ± 1.0 | 2 | 0.8 ± 1.0 | 0 | — |
| Control | 4 | — | — | — | |||
| Week after DSS administration | No. of rats analyzed | Dysplastic foci | Adenoma | Adenocarcinoma | |||
| Incidence | Number/rat | Incidence | Number/rat | Incidence | Number/rat | ||
| 1 | 4 | 0 | — | 0 | — | 0 | — |
| 4 | 4 | 1 | 0.3 ± 0.1 | 0 | — | 0 | — |
| 6 | 4 | 3 | 1.8 ± 1.7 | 1 | 0.3 ± 0.1 | 1 | 0.3 ± 0.1 |
| 8 | 4 | 2 | 0.8 ± 1.0 | 2 | 0.8 ± 1.0 | 0 | — |
| Control | 4 | — | — | — | |||
Mean ± SD.
The values are based on findings with longitudinally embedded sections.
Immunohistochemistry and cell proliferative activity
Immunoreactivity of β-catenin was found in the epithelial cells ( Figure 2B ) with weak reactions in the endothelium of blood vessels and ganglion cells in submucosal and myenteric (Meissner's and Auerbach's) plexus in non-treated control rats. No obvious aberrant expression of β-catenin in mucosal epithelium was detected at week 1 in DMH–DSS-treated animals ( Figure 1C ). At weeks 4–8, nuclear/cytoplasmic expression of β-catenin was exhibited in subsets of epithelial cells in the regenerative mucosa with Paneth cell metaplasia ( Figure 2H and 3C ) but this was not evident in tissue without Paneth cells ( Figure 2E ). The β-catenin-accumulated epithelial cells with and without Paneth cell metaplasia were frequently located not only in the lower crypt zone ( Figure 2H ) but also in laterally branching crypts in the middle zone ( Figure 3C ). Accumulation of β-catenin was also detected in the dysplastic foci, adenomas and adenocarcinomas, with higher levels of expression than in the regenerative mucosa with Paneth cells ( Figure 3C ). Ki-67 positivity of epithelial cells in the reactive hyperplastic mucosa was significantly ( P < 0.01) greater than that in normal mucosa in the controls and normal-appearing mucosa in the DMH–DSS-treated animals at week 1 ( Figure 1D and 2C , Table III ), whereas values for regenerative mucosa with and without Paneth cell metaplasia and β-catenin accumulation were similar, and significantly ( P < 0.05) lower than in normal-appearing mucosa at weeks 4–6 ( Figure 2F and I , Table III ). Ki-67-positive cells were mainly localized in the lower crypt zone of the regenerative mucosa without Paneth cell metaplasia, but with Paneth cell metaplasia the Ki-67-positive cells were detected not only in the lower zone but also in the middle zone of the regenerative mucosa.
Cell proliferative activity in reactive hyperplastic mucosa and regenerative mucosa in the distal colon/rectum of rats treated with DSS after DMH initiation
| Week after DSS administration | Type of lesion a | No. of rats analyzed | Ki-67 positivity b (%) |
| 1 | Normal-appearing mucosa | 4 | 38.7 ± 12.4 |
| Reactive hyperplastic mucosa | 4 | 73.2 ± 12.7 c | |
| 4–6 | Normal-appearing mucosa | 8 | 34.4 ± 9.3 |
| RM with Paneth cell metaplasia/β-catenin accumulation | 8 | 25.9 ± 8.4 d | |
| RM without Paneth cell metaplasia/β-catenin accumulation | 7 | 23.3 ± 7.3 d | |
| Control | Normal mucosa | 4 | 38.9 ± 12.6 |
| Week after DSS administration | Type of lesion a | No. of rats analyzed | Ki-67 positivity b (%) |
| 1 | Normal-appearing mucosa | 4 | 38.7 ± 12.4 |
| Reactive hyperplastic mucosa | 4 | 73.2 ± 12.7 c | |
| 4–6 | Normal-appearing mucosa | 8 | 34.4 ± 9.3 |
| RM with Paneth cell metaplasia/β-catenin accumulation | 8 | 25.9 ± 8.4 d | |
| RM without Paneth cell metaplasia/β-catenin accumulation | 7 | 23.3 ± 7.3 d | |
| Control | Normal mucosa | 4 | 38.9 ± 12.6 |
RM, regenerative mucosa.
Mean ± SD.
P < 0.01.
P < 0.05 versus corresponding normal-appearing mucosa.
The values are based on findings with longitudinally embedded sections.
Cell proliferative activity in reactive hyperplastic mucosa and regenerative mucosa in the distal colon/rectum of rats treated with DSS after DMH initiation
| Week after DSS administration | Type of lesion a | No. of rats analyzed | Ki-67 positivity b (%) |
| 1 | Normal-appearing mucosa | 4 | 38.7 ± 12.4 |
| Reactive hyperplastic mucosa | 4 | 73.2 ± 12.7 c | |
| 4–6 | Normal-appearing mucosa | 8 | 34.4 ± 9.3 |
| RM with Paneth cell metaplasia/β-catenin accumulation | 8 | 25.9 ± 8.4 d | |
| RM without Paneth cell metaplasia/β-catenin accumulation | 7 | 23.3 ± 7.3 d | |
| Control | Normal mucosa | 4 | 38.9 ± 12.6 |
| Week after DSS administration | Type of lesion a | No. of rats analyzed | Ki-67 positivity b (%) |
| 1 | Normal-appearing mucosa | 4 | 38.7 ± 12.4 |
| Reactive hyperplastic mucosa | 4 | 73.2 ± 12.7 c | |
| 4–6 | Normal-appearing mucosa | 8 | 34.4 ± 9.3 |
| RM with Paneth cell metaplasia/β-catenin accumulation | 8 | 25.9 ± 8.4 d | |
| RM without Paneth cell metaplasia/β-catenin accumulation | 7 | 23.3 ± 7.3 d | |
| Control | Normal mucosa | 4 | 38.9 ± 12.6 |
RM, regenerative mucosa.
Mean ± SD.
P < 0.01.
P < 0.05 versus corresponding normal-appearing mucosa.
The values are based on findings with longitudinally embedded sections.
Mutations of the β-catenin gene in regenerative and neoplastic lesions
Twenty-seven regenerative lesions with Paneth cell metaplasia, five dysplastic foci with Paneth cells, one dysplastic focus without Paneth cells, two adenomas and one adenocarcinoma with Paneth cells could be subjected to PCR-based direct sequencing for the β-catenin gene. Six of 27 (22%) regenerative and two of six (33%) dysplastic foci and one adenoma and carcinoma each were found to harbor point mutations. Data for mutation status for phosphorylation sites and mutation hot spots of β-catenin gene are summarized in Table IV . Mutations at codons 34 and 41 were the most common, and all mutations found in the lesions caused amino acid substitution. No significant correlation between the mutation status and immunohistochemical intensity for β-catenin was observed across lesions.
Mutation status of the β- catenin in regenerative mucosa samples with Paneth cell metaplasia and pre-neoplastic and neoplastic lesions in the distal colon and rectal mucosa of rats treated with DSS after DMH initiation
| Sample | Week after DSS administration | Immunohistochemical intensity for β-catenin a | Mutation status | ||
| Codon | Type of mutation | AA substitution | |||
| RM | 4 | + | 34 | GGA→GAA | Gly→Glu |
| RM | 4 | + | 34 | GGA→GAA | Gly→Glu |
| RM | 4 | ++ | Wild type | ||
| RM | 4 | ++ | Wild type | ||
| RM | 4 | + | Wild type | ||
| RM | 4 | + | Wild type | ||
| RM | 4 | + | Wild type | ||
| RM | 4 | + | Wild type | ||
| RM | 4 | + | Wild type | ||
| RM | 6 | ++ | 33 | TCT→TGT | Ser→Cys |
| RM | 6 | + | 34 | GGA→GAA | Gly→Glu |
| RM | 6 | + | 41 | ACC→ATC | Thr→Ile |
| RM | 6 | ++ | Wild type | ||
| RM | 6 | ++ | Wild type | ||
| RM | 6 | ++ | Wild type | ||
| RM | 6 | ++ | Wild type | ||
| RM | 6 | + | Wild type | ||
| RM | 6 | + | Wild type | ||
| RM | 6 | + | Wild type | ||
| RM | 6 | + | Wild type | ||
| RM | 6 | + | Wild type | ||
| RM | 6 | + | Wild type | ||
| RM | 6 | + | Wild type | ||
| RM | 6 | + | Wild type | ||
| RM | 8 | + | 41 | ACC→ATC | Thr→Ile |
| RM | 8 | ++ | Wild type | ||
| RM | 8 | ++ | Wild type | ||
| Dy | 4 | + | Wild type | ||
| Dy | 6 | + | 34 | GGA→GAA | Gly→Glu |
| Dy b | 6 | + | 41 | ACC→ATC | Thr→Ile |
| Dy | 6 | ++ | Wild type | ||
| Dy | 8 | ++ | Wild type | ||
| Dy | 8 | ++ | Wild type | ||
| Ad | 8 | ++ | 41 | ACC→ATC | Thr→Ile |
| Ad | 8 | ++ | Wild type | ||
| Ca | 6 | ++ | 37 | TCT→TTT | Ser→Ile |
| Sample | Week after DSS administration | Immunohistochemical intensity for β-catenin a | Mutation status | ||
| Codon | Type of mutation | AA substitution | |||
| RM | 4 | + | 34 | GGA→GAA | Gly→Glu |
| RM | 4 | + | 34 | GGA→GAA | Gly→Glu |
| RM | 4 | ++ | Wild type | ||
| RM | 4 | ++ | Wild type | ||
| RM | 4 | + | Wild type | ||
| RM | 4 | + | Wild type | ||
| RM | 4 | + | Wild type | ||
| RM | 4 | + | Wild type | ||
| RM | 4 | + | Wild type | ||
| RM | 6 | ++ | 33 | TCT→TGT | Ser→Cys |
| RM | 6 | + | 34 | GGA→GAA | Gly→Glu |
| RM | 6 | + | 41 | ACC→ATC | Thr→Ile |
| RM | 6 | ++ | Wild type | ||
| RM | 6 | ++ | Wild type | ||
| RM | 6 | ++ | Wild type | ||
| RM | 6 | ++ | Wild type | ||
| RM | 6 | + | Wild type | ||
| RM | 6 | + | Wild type | ||
| RM | 6 | + | Wild type | ||
| RM | 6 | + | Wild type | ||
| RM | 6 | + | Wild type | ||
| RM | 6 | + | Wild type | ||
| RM | 6 | + | Wild type | ||
| RM | 6 | + | Wild type | ||
| RM | 8 | + | 41 | ACC→ATC | Thr→Ile |
| RM | 8 | ++ | Wild type | ||
| RM | 8 | ++ | Wild type | ||
| Dy | 4 | + | Wild type | ||
| Dy | 6 | + | 34 | GGA→GAA | Gly→Glu |
| Dy b | 6 | + | 41 | ACC→ATC | Thr→Ile |
| Dy | 6 | ++ | Wild type | ||
| Dy | 8 | ++ | Wild type | ||
| Dy | 8 | ++ | Wild type | ||
| Ad | 8 | ++ | 41 | ACC→ATC | Thr→Ile |
| Ad | 8 | ++ | Wild type | ||
| Ca | 6 | ++ | 37 | TCT→TTT | Ser→Ile |
RM, regenerative mucosa with Paneth cell metaplasia; Dy, dysplastic focus; Ad, adenoma; Ca, adenocarcinoma.
+, weak to moderate; ++, strong.
Dysplasia without Paneth cell metaplasia.
Mutation status of the β- catenin in regenerative mucosa samples with Paneth cell metaplasia and pre-neoplastic and neoplastic lesions in the distal colon and rectal mucosa of rats treated with DSS after DMH initiation
| Sample | Week after DSS administration | Immunohistochemical intensity for β-catenin a | Mutation status | ||
| Codon | Type of mutation | AA substitution | |||
| RM | 4 | + | 34 | GGA→GAA | Gly→Glu |
| RM | 4 | + | 34 | GGA→GAA | Gly→Glu |
| RM | 4 | ++ | Wild type | ||
| RM | 4 | ++ | Wild type | ||
| RM | 4 | + | Wild type | ||
| RM | 4 | + | Wild type | ||
| RM | 4 | + | Wild type | ||
| RM | 4 | + | Wild type | ||
| RM | 4 | + | Wild type | ||
| RM | 6 | ++ | 33 | TCT→TGT | Ser→Cys |
| RM | 6 | + | 34 | GGA→GAA | Gly→Glu |
| RM | 6 | + | 41 | ACC→ATC | Thr→Ile |
| RM | 6 | ++ | Wild type | ||
| RM | 6 | ++ | Wild type | ||
| RM | 6 | ++ | Wild type | ||
| RM | 6 | ++ | Wild type | ||
| RM | 6 | + | Wild type | ||
| RM | 6 | + | Wild type | ||
| RM | 6 | + | Wild type | ||
| RM | 6 | + | Wild type | ||
| RM | 6 | + | Wild type | ||
| RM | 6 | + | Wild type | ||
| RM | 6 | + | Wild type | ||
| RM | 6 | + | Wild type | ||
| RM | 8 | + | 41 | ACC→ATC | Thr→Ile |
| RM | 8 | ++ | Wild type | ||
| RM | 8 | ++ | Wild type | ||
| Dy | 4 | + | Wild type | ||
| Dy | 6 | + | 34 | GGA→GAA | Gly→Glu |
| Dy b | 6 | + | 41 | ACC→ATC | Thr→Ile |
| Dy | 6 | ++ | Wild type | ||
| Dy | 8 | ++ | Wild type | ||
| Dy | 8 | ++ | Wild type | ||
| Ad | 8 | ++ | 41 | ACC→ATC | Thr→Ile |
| Ad | 8 | ++ | Wild type | ||
| Ca | 6 | ++ | 37 | TCT→TTT | Ser→Ile |
| Sample | Week after DSS administration | Immunohistochemical intensity for β-catenin a | Mutation status | ||
| Codon | Type of mutation | AA substitution | |||
| RM | 4 | + | 34 | GGA→GAA | Gly→Glu |
| RM | 4 | + | 34 | GGA→GAA | Gly→Glu |
| RM | 4 | ++ | Wild type | ||
| RM | 4 | ++ | Wild type | ||
| RM | 4 | + | Wild type | ||
| RM | 4 | + | Wild type | ||
| RM | 4 | + | Wild type | ||
| RM | 4 | + | Wild type | ||
| RM | 4 | + | Wild type | ||
| RM | 6 | ++ | 33 | TCT→TGT | Ser→Cys |
| RM | 6 | + | 34 | GGA→GAA | Gly→Glu |
| RM | 6 | + | 41 | ACC→ATC | Thr→Ile |
| RM | 6 | ++ | Wild type | ||
| RM | 6 | ++ | Wild type | ||
| RM | 6 | ++ | Wild type | ||
| RM | 6 | ++ | Wild type | ||
| RM | 6 | + | Wild type | ||
| RM | 6 | + | Wild type | ||
| RM | 6 | + | Wild type | ||
| RM | 6 | + | Wild type | ||
| RM | 6 | + | Wild type | ||
| RM | 6 | + | Wild type | ||
| RM | 6 | + | Wild type | ||
| RM | 6 | + | Wild type | ||
| RM | 8 | + | 41 | ACC→ATC | Thr→Ile |
| RM | 8 | ++ | Wild type | ||
| RM | 8 | ++ | Wild type | ||
| Dy | 4 | + | Wild type | ||
| Dy | 6 | + | 34 | GGA→GAA | Gly→Glu |
| Dy b | 6 | + | 41 | ACC→ATC | Thr→Ile |
| Dy | 6 | ++ | Wild type | ||
| Dy | 8 | ++ | Wild type | ||
| Dy | 8 | ++ | Wild type | ||
| Ad | 8 | ++ | 41 | ACC→ATC | Thr→Ile |
| Ad | 8 | ++ | Wild type | ||
| Ca | 6 | ++ | 37 | TCT→TTT | Ser→Ile |
RM, regenerative mucosa with Paneth cell metaplasia; Dy, dysplastic focus; Ad, adenoma; Ca, adenocarcinoma.
+, weak to moderate; ++, strong.
Dysplasia without Paneth cell metaplasia.
Discussion
In the present study, distal parts of the colon and rectal mucosa were found to undergo extensive loss of crypts along with inflammatory cell infiltration and neighboring reactive hyperplasia in rats after 1 week DSS treatment following DMH initiation. Repair was evident within several weeks with the appearance of regenerative mucosa having an abnormal architecture but lacking distinct cellular atypia. In the regenerative mucosa, particularly in regions with inflammatory change, Paneth cell metaplasia was additionally characterized. In quiescent phase or long-standing cases of IBD patients, similar histopathological changes including crypt distortion, mucosal atrophy and Paneth cell metaplasia have been described ( 7 ). In addition, Paneth cell metaplasia is particularly numerous in patients with long-standing colitis who develop malignant change ( 7 ), although the underlying etiology of the Paneth cell metaplasia in response to injury has yet to be clarified ( 43 ). Whether the re-epithelialization of large patches of colonic mucosa by abnormal clones is simply a consequence of the healing response to ulceration caused by chronic inflammation or the epithelial cells of IBD patients have an innate ability to replace surrounding epithelium is also unknown ( 2 ). Both DMH-induced tumors and DSS-induced mucosal damaged/reactive changes are more prevalent in the distal part of the colon/rectum in rats ( 44 , 45 ), suggesting promoting effects of DSS-associated mucosal lesions on DMH-induced carcinogenesis to be limited to the affected tissue. In our previous experiment, Paneth cell metaplasia in non-dysplastic/neoplastic colorectal mucosa did not appear in rats treated with DMH or DSS alone ( 39 ). Therefore, the cause of Paneth cell metaplasia induced within several weeks after DSS treatment in the present rat model, in contrast to long-standing IBD cases, remains unknown, but DMH initiation is presumbly associated with the differentiation abnormality which is only likely to occur in rats treated with DMH and DSS. Most dysplastic foci were found in regions with Paneth cells, and the fact that dysplastic and neoplastic lesions also frequently showed Paneth cell differentiation suggests that substantial genomic alterations associated with DSS-induced mucosal damage can occur and contribute to carcinogenesis.
A striking feature in the present study was the alteration in subcellular localization of β-catenin exhibited by subsets of epithelial cells in the regenerative mucosa with Paneth cells. Point mutations of β-catenin gene were also detected in 6 of 27 regenerative mucosa samples with Paneth cells. Thus, accumulation of β-catenin might have been caused, at least partly, by gene alterations occurring in the regenerative colorectal mucosa with Paneth cells in the DMH–DSS rats. In our previous experiment, β-catenin accumulation in non-dysplastic/neoplastic colorectal mucosal lesions was not evident in rats treated with DMH or DSS alone ( 39 ). Although β-catenin mutations have previously been found in early dysplastic lesions in rat colorectal carcinogenesis induced by genotoxic carcinogens ( 34 , 36 , 44 ), to our knowledge, this is the first report of their demonstration in non-dysplastic tissue after mucosal damage in rodent colorectal carcinogenesis. In previous literature, β-catenin gene mutations, most commonly at codon 32, were demonstrated in early dysplastic lesions and tumors induced by DMH ( 44 ), and another report showed the mutation cluster around codon 33 in DMH-induced tumors in the rat colon ( 46 ). With the non-dysplastic regenerated mucosa with Paneth cells, dysplastic foci and tumors analyzed in the present study, β-catenin mutations at codons 34 and 41 were the most common. Interestingly, the mutated codon 41 directly substitutes a critical threonine phosphorylation site ( 47 ) and has been found in human colon tumors with wild-type APC ( 11 ). Yamada et al. have demonstrated β-catenin mutations to be scattered in exon 3 of the gene in early dysplastic lesions, while they are converged at codons encoding functionally important residues in tumors in azoxymethane-induced rat colon carcinogenesis. This indicates β-catenin mutations to be selected during malignant transformation ( 48 ). On the other hand, Blum et al. showed that the spectrum of β-catenin mutations in carcinogen-induced rat colon tumors can be altered from a cluster around Ser33 to codons 37, 41 and 45 by post-initiation exposure to a phytochemicals. Thus, we can speculate the existence of a survival advantage for cells containing mutant forms of β-catenin with substitutions in Ser37, Thr41 or Ser45 ( 46 ). The present results are clearly in line with DSS-associated mucosal damage causing cell proliferation in reactive hyperplastic mucosa and acceleration of such mutated gene selection to promote rat colorectal carcinogenesis.
Neoplastic transformation and progression are known to be associated with the release of highly reactive oxygen and nitrogen species and/or cytokines from inflammatory cells ( 2 , 49 ) and the regenerative mucosa with Paneth cells was significantly linked to interstitial inflammation in the present study. Therefore, inflammatory changes not only in the early mucosal damaged condition but also in the subsequent duration with mucosal repair and regeneration under Paneth cell inducing conditions might also play roles. The cause of the decreased proportion of regenerative mucosa with Paneth cells and β-catenin accumulation at later time points, suggesting the aberrant phenotype in regenerative mucosa to be partly reversible, is unclear. Although subsets of β-catenin-accumulated epithelial cells might have pre-neoplastic potential, there is possibility that other more ‘normal’ subsets are easily affected by cytotoxic inflammatory cells.
No significant correlation between the mutation status and immunohistochemical intensity for β-catenin in the lesions was observed, suggesting β-catenin mutations might not be sufficient in themselves for accumulation of the protein. A similar phenomenon has been demonstrated in pre-neoplastic and neoplastic lesions in the colons of carcinogen-treated rats ( 36 , 46 ). Further study is needed to clarify the existence of alterations in other related factors within the APC/β-catenin/Tcf signal transduction pathway. β-Catenin regulates expression of growth-promoting proteins, such as c-myc ( 41 ) and cyclin D1 ( 42 ), and increased β-catenin expression is accompanied by nuclear translocation in murine colon epithelium ( 50 ). Therefore, the accumulation of β-catenin in the regenerative mucosa with Paneth cells might indicate dysfunction of cell proliferation regulatory signals and potential oncogenesis. Wnt signaling, which is transduced through β-catenin/Tcf-4 in intestinal crypts, has recently been reported to drive a Paneth cell maturation program ( 51 ). In addition, Andreu et al. ( 52 ) demonstrated that activation of the β-catenin signaling pathway following APC loss promoted differentiation along the Paneth cell lineage in small intestinal epithelial cells in a transgenic mouse model. Accumulation of β-catenin associated with gene mutations might therefore be causally linked with the Paneth cell differentiation of the epithelial cells in the regenerative mucosa and neoplastic lesions in the present rat model. This conclusion is not necessarily inconsistent with the presence of one dysplastic focus without Paneth cell metaplasia containing a mutation at codon 41, since enhanced β-catenin signaling does not appear to be sufficient for the complete differentiation of Paneth cells, as identified by the presence of typical secretory granules ( 53 , 54 ).
In conclusion, regenerative mucosa with Paneth cells, β-catenin accumulation and interstitial inflammatory cell infiltration is possibly involved in the acceleration of colorectal carcinogenesis in this DMH–DSS rat model. The model resembles human IBD cases regarding inflammation-associated Paneth cell metaplasia, but β-catenin accumulation is not necessarily consistent with the human situation. Epithelial regeneration accompanied by chronic inflammation, thus, might induce non-genetic alteration as well as gene mutations and mutated gene selection as a part of the oncogenic events in both humans and experimental animals.
Funding
The Grant-in-Aid for Cancer Research (17S-6) from the Ministry of Health, Labour and Welfare of Japan.
Abbreviations
- DMH
1,2-dimethylhydrazine
- DSS
dextran sodium sulfate
- IBD
inflammatory bowel disease
- PCR
polymerase chain reaction
- UC
ulcerative colitis
We thank Ms Ayako Kaneko for her expert technical assistance.
Conflict of Interest statement: None declared.

{kind=link}
{kind=link}
{kind=link}