




Abstract
Compelling evidences have rendered the tumor microenvironment a crucial determinant in cancer outcome. Activating transcription factor 3 (ATF3), a stress response transcription factor, is known to have a dichotomous role in tumor cells, acting either as a tumor suppressor or an oncogene in a context-dependent manner. However, its expression and possible role in the tumor microenvironment are hitherto unknown. Here we show that ATF3 is upregulated in the stromal compartment of several types of cancer. Accordingly, Cancer-associated fibroblasts (CAFs) ectopically expressing ATF3 proliferated faster as indicated by increased colony-forming capacity and promoted the growth of adjacent tumor cells when co-injected into nude mice. Utilizing a genome-wide profiling approach, we unraveled a robust gene expression program induced by ATF3 in CAFs. Focusing on a specific subset of genes, we found that the ability of stromal ATF3 to promote cancer progression is mediated by transcriptional repression of CLDN1 and induction of CXCL12 and RGS4 . In addition, regulation of LIF , CLDN1 , SERPINE2 , HSD17B2 , ITGA7 and PODXL by ATF3 mediated the increased proliferation capacity of CAFs. In sum, our findings implicate ATF3 as a novel stromal tumor promoter and suggest that targeting ATF3 pathway might be beneficial for anticancer therapy.
Introduction
In recent years, a growing body of evidence has shaped the notion that tumorigenesis is governed by a complex network of tumor cells, stromal residents such as fibroblasts, endothelial cell and infiltrating immune cells and the extracellular matrix ( 1–3 ). The cancer-promoting communication between tumor cells and their surrounding stroma escalates throughout the transformation process and has been termed the ‘vicious cycle’ ( 4 ). Anticancer drugs, specifically designed to target this interaction, are being developed, and an increasing number of such drugs is currently under clinical trials ( 5–7 ). It is therefore crucial to identify key players that operate in the stroma and to decipher the mechanism by which they exert their tumorigenic effect.
The activating transcription factor 3 ( ATF3 ) gene encodes a member of the ATF/cyclic adenosine monophosphate response element-binding family of transcription factors, which share the basic region/leucine zipper DNA-binding motif and bind to the CREB/ATF and AP-1 consensus sequences ( 8 ). ATF3 is a stress response protein, which in normal cells responds to a plethora of insults such as Endoplasmic Reticulum stress, DNA damage, tissue injury and metabolic stress and either through induction or repression of numerous targets executes cell fate decisions such as cell cycle arrest or apoptosis ( 9–14 ).
During tumorigenesis, ATF3 might promote cancer or hinder its progression depending on the tumor type and its genetic makeup ( 15 ). However, a possible role for ATF3 in the tumor microenvironment has not been proposed, although several studies have shown that ATF3 might affect potential members of the stromal milieu such as endothelial cells, smooth muscle cells and macrophages. For example, ATF3 was shown to protect endothelial cells from tumor necrosis factor α-induced cell death ( 16 ). Furthermore, ATF3 induction in endothelial cells endowed them with the ability to form tubule structures ( 17 ). A more recent study revealed that ATF3 increases the survival and enhances the migration of vascular smooth muscle cells ( 18 ). These findings imply that ATF3 might be involved in cancer progression through the ability to promote the survival and formation of blood vessels in the vicinity of the tumor. ATF3 was also extensively studied in the context of the innate immune response regulation ( 15 ). Accordingly, it was shown to be a transcriptional regulator of Toll Like Receptors signaling in macrophages ( 19 , 20 ). The fact that ATF3 restrains the immune response might be utilized by the adjacent tumor cells to evade immunosurveillance.
In light of these findings, the primary motivation of this work was to investigate the possible role of ATF3 in the stromal compartment of tumors. We found that ATF3, which is kept at low levels in normal cells, is highly expressed in the stromal compartment of various cancers. Our findings suggest that stromal ATF3 assumes an active role in cancer progression by (i) granting stromal cells with a growth advantage via transcriptional regulation of LIF , CLDN1 , SERPINE2 , HSD17B2 , ITGA7 and PODXL and by (ii) enhancing tumor cell growth through transcriptional repression of CLDN1 and activation of CXCL12 and RGS4 .
Materials and methods
Cell lines
Cancer-associated fibroblasts (CAFs) from prostate, designated PF-179, and breast and lung CAFs designated ZB-06 and HK-3, respectively (a kind gift from Dr Jair Bar) were isolated from the stromal compartments of tumor specimens and grown in culture. The letter ‘T’ is used to denote that the designated cell line was immortalized with the human telomerase catalytic subunit (hTERT). CAFs and WI-38 human embryonic lung fibroblasts were grown in minimum essential medium supplemented with 10% fetal bovine serum, 2 mM l -glutamine, 1 mM sodium pyruvate and antibiotics. Cancer cell lines (MDA-231, MDA-468, MCF-7, SKBR-3, SW-480, HT-29, HCT-116, HepG-2, PC-3, LNCaP, DU-145, H-460, H-1299, NHBET and HT-1080) were grown according to ATCC protocols. Cells were maintained in a humidified incubator at 37°C and 5% CO 2 .
Tissue arrays
Two commercial cancer tissue arrays (MC2081 and MC803, US-Biomax, Rockville, MD) consisting of biopsies from several types of cancer (colon, rectum, breast, prostate, lung, retinoblastoma, nephroblastoma and neuroblastoma), as well as a set of biopsies from 107 lung cancers (Sheba Medical Center), were stained with an anti-ATF3 antibody (HPA001562, Sigma, Rehovot, Israel) as described below. MC2081 was also stained with an anti-CXCL12 antibody (MAB350, R&D Systems, Minneapolis, MN)
Sarcomas samples
RNA samples were isolated from various human sarcoma specimens. Lipo-sarcomas (5) and various sarcomas as follows: fibrosarcomas (3) , leiomyosarcoma (5) , neurofibrosarcoma, dermatofibrosarcoma and rhabdomyosarcoma. Samples were kindly provided by Dr Paul Meltzer (National Institutes of Health, Bethesda, MD).
Subcutaneous injections of tumor cells
All in vivo experiments were reviewed and approved by the Institutional Animal Care and Use Committee of the Weizmann institute (Permit numbers: 80109-2 and 310110-1). Ten immunodeficient mice (CD-1-Hfh11 nu Harlan Laboratories, Rehovot, Israel) were anesthetized with a mixture of 100 mg/kg ketamine and 10 mg/kg xylazin (Ketaset). Cells (2 × 10 6 /50 μl per injection, in phosphate-buffered saline) were then subcutaneously injected into each hind limb using a 30 gauge needle and a 0.1 ml syringe. Mice were sacrificed when tumor volume reached 1 cm 3 . Tumors were then removed, photographed and weighed. For co-injection experiments, the same setup was used with a mixture of 2 × 10 6 fibroblasts and 1 × 10 6 tumor cells/50 μl phosphate-buffered saline.
Western blot analysis
Total cell extracts were fractionated by gel electrophoresis; proteins were transferred to nitrocellulose membranes and immunoblotted with Rabbit Polyclonal Anti-ATF3 (Sc-188, Santa Cruz Biotechnology, Santa Cruz, California) and anti-GAPDH (MAB374; Chemicon, Billerica, MA). The protein–antibody complexes were detected by horseradish peroxidase-conjugated secondary antibodies followed by the enhanced SuperSignal west pico chemiluminescent substrate (Thermo Scientific, Huntsville, IL).
Genes overexpression
Overexpression plasmids were constructed by cloning the desired open reading frames using reverse transcription–polymerase chain reaction (PCR) with specific primers (see Supplementary Materials and methods , available at Carcinogenesis Online). Products were transferred into the pGEMT-easy vector (Promega, Madison, WI) and then restricted with EcoRI and inserted into either pBABE-Neo or PLXSN-Neo-expressing vectors. Cells were then retrovirally infected as described elsewhere ( 21 ).
Gene knockdown
Expression of the specified genes was knocked down using Human pLKO.1 lentiviral short hairpin RNA target gene sets (Open Biosystems, Thermo Fisher Scientific, Huntsville, AL) according to the manufacturer protocol and was compared with an empty vector.
Isolation of total RNA and quantitative real-time PCR
Total RNA was isolated using the RNAeasy kit (Qiagen) according to the manufacturer’s protocol. A 2 μg aliquot was reverse transcribed using MMLV-RT (Bio-RT) and random hexamer primers. Quantitative real-time PCR was performed on an ABI 7300 instrument (Applied Biosystems, Foster City, CA) using Platinum SYBR Green and qPCR SuperMix (Invitrogen, Carlsbad, CA).
Microarray
Total RNA was extracted using Tri-Reagent (MRC, London, England) according to manufacturer’s protocol and sent to the MicroArray unit (Weizmann Institute of Science, Rehovot, Israel). The procedure was performed using Affymetrix HU-GENE1.0st oligonucleotide arrays with technical duplicates. Raw analyses were performed using BRB-ArrayTools developed by Dr Richard Simon and BRB-ArrayTools development team ( 22 ). For ATF3 differential expression, a class comparison analysis was performed, comparing ATF3-expressing cells to empty control and ATF3ΔZip, with a threshold of P < 0.05 and log 1.2-fold change.
Colony-forming efficiency assay
Cells were seeded in triplicates at a cell density of 20 cells per square centimeter. Medium was replaced every 4 days. Two weeks after plating, plates were stained with crystal violet and scanned and colonies were counted.
Immunostaining
Formalin-fixed and paraffin-embedded sections of lung tissue microarray blocks, 4 μm thick, were dewaxed in xylene and rehydrated. Antigen retrieval was performed using a pressure cooker (Milestone, Microwave Laboratory Systems, Peine, Germany) at 120°C for 5 min in citrate buffer, pH 6, and slides were rinsed with Tris-buffered saline buffer. Subsequently, an endogenous peroxidase block was performed for 10 min in 3% H 2 O 2 /phosphate-buffered saline. After Tris-buffered saline rinses, sections were blocked with 10% goat serum for 30 min at room temperature and subsequently incubated with the relevant primary antibody (ATF3, CXCL12 described above or Ki-67—MU297-UC, Biogenex, San Ramon, CA) overnight at 4°C. Detection was performed with the Histostain SP Broad Spectrum kit (Zymed Laboratories, San Francisco, CA, Invitrogen, Carlsbad, CA). Briefly, sections were incubated with a biotinylated secondary antibody and subsequently, after Tris-buffered saline rinse, with horseradish peroxidase–streptavidin for 30 min at room temperature. Visualization was performed with the substrate–chromogen AEC, counterstained with hematoxylin and coverslipped with an aqueous mounting fluid (glycergel).
Transfections and reporter assays
HT-1080 cells were plated at a density of 2 × 10 4 cells per well in a six well plate for 24 h prior to transfection. Cells were then transfected using FuGene HD (Roche, Mannheim, Germany) with 1 microgram per well of CXCL12 promoter–luciferase reporter construct and 400 nanograms per well pLXSN-Renilla expression vector for normalization of transfection efficiency. Cell extracts were prepared with passive lysis buffer (Promega) and luciferase and Renilla activities were determined using a Victor luminometer (Wallac, Waltham, MA).
Statistical analysis
Unless stated otherwise, an unpaired one-tailed student’s t -test was performed; the resulted P -value is shown. All quantitative real-time PCR experiments were repeated at least three times, and the results were normalized to the expression of GAPDH and presented as a mean ± standard deviation of two duplicate runs from a typical experiment.
Chromatin immunoprecipitation
Chromatin immunoprecipitation was conducted as described ( 23 ).
Primers list
Specific primers for quantitative real-time PCR, Chromatin immunoprecipitation and overexpression are listed in the Supplementary Materials and methods (available at Carcinogenesis Online).
Results
ATF3 is upregulated in the stromal compartment of human tumors
ATF3 expression was reported previously to be elevated in tumors ( 24 , 25 ). Accordingly, we found ATF3 to be upregulated both at the messenger RNA (mRNA) and the protein levels in diverse types of tumors ( Supplementary Figure S1A –D is available at Carcinogenesis Online), whereas its levels in normal tissues were low. Also in agreement with previous studies ( 25–31 ), modulation of ATF3 in various cancer cell lines had cell-specific effects, ranging from suppression to promotion of tumor growth in vivo ( Supplementary Table S1 and Figure S1E and F are available at Carcinogenesis Online). Interestingly, in addition to the frequent overexpression of ATF3 in primary tumors, elevation of ATF3 protein levels was also evident in the tumor microenvironment of the colon, lung and breast cancer samples ( Figure 1A , right panel), with a frequency of 46, 25 and 29% positive biopsies, respectively. The colon and lung samples exhibited both nuclear and cytoplasmatic expression of stromal ATF3, whereas the breast samples showed a cytoplasmatic and a more diffusible expression pattern. In contrast, only a small portion of the rectum and prostate specimens had positive staining for stromal ATF3. As a control, a corresponding non-malignant tissue located 1.5 cm from the tumor was stained and revealed no traces of ATF3 in the stromal compartment ( Figure 1A , left panel). Moreover, as depicted in Figure 1B and C , ATF3 was upregulated in CAFs dissected from a prostatic tumor margins compared with distal fibroblasts (normal-associated fibroblasts, NAFs). Furthermore, ATF3 levels were significantly elevated in cancer-associated stroma removed from breast cancer patients compared with normal stroma, in a data set retrieved from the ‘Ocnomine’ database ( 32 , 33 ) ( Figure 1D ).
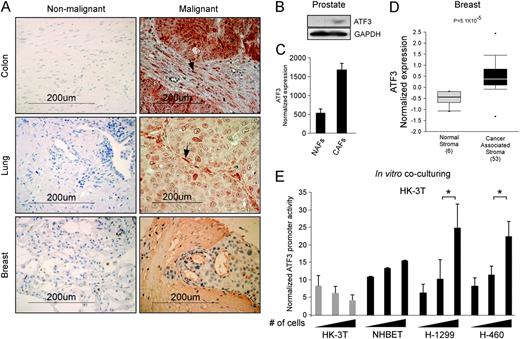
ATF3 expression in the tumor microenvironment. ( A ) (Right panel) Cancer samples from colon, lung and breast cancer patients were stained with an anti-ATF3 antibody and photographed. Arrows point at stromal cells with high ATF3 staining. (Left panel) Non-malignant tissue located 1.5 cm away from the tumor served as a control for the corresponding tumor type. ( B ) NAFs and CAFs originating from prostate cancer ( 38 ) were analyzed for ATF3 expression by (B) western blot and ( C ) quantitative real-time PCR, P = 0.001. ( D ) The Oncomine database was queried for studies in which ATF3 expression significantly differs between NAFs and breast CAFs ( 32 , 33 ). The P -value designates the probability that the observed difference in ATF3 expression between normal and cancer samples is due to random chance. The bars represent 25–75% interval of normalized ATF3 expression levels in each group. The deviation lines represent 10–90% percentiles, and dots represent samples with minimum and maximum expression level in a given group. ( E ) Lung fibroblasts (HK-3T) expressing a luciferase gene downstream of ATF3 promoter were cocultured with growing amounts of fibroblasts, normal lung bronchial cells (NHBET) or the lung cancer cell lines H-460 and H-1299. ATF3 promoter activity is indicated by normalized luciferase luminescence intensity. * P < 0.05.
Since ATF3 is upregulated in the vicinity of tumors, we reasoned that cancer cells might benefit from the expression of ATF3 in the stroma and thus induce its expression in the stromal compartment. To address this question, we cloned ATF3 promoter into a retroviral luciferase reporter plasmid and inserted it into immortalized human lung CAFs (HK-3T). Subsequently, the fibroblasts were cocultivated with an increasing number of human lung cancer cell lines, either H-1299 or H-460, or with normal lung bronchial cells and non-infected fibroblasts as a control. ATF3 promoter activity was elevated in a cell number-dependent manner in the fibroblasts when cocultivated with both lung cancer cell lines, but not with the controls—fibroblasts or normal lung cells ( Figure 1E ). Combined, these results demonstrate that signals communicated from the cancer cells to their stroma can induce stromal ATF3 transcription.
Ectopic expression of ATF3 in CAFs promotes their proliferation and induces the growth of adjacent tumor cells
To evaluate the impact of stromal ATF3 expression, we ectopically expressed its coding sequence in three different types of fibroblasts, originating from prostate, breast and lung tumors, as well as in the WI-38 lung immortalized fibroblasts, which often serve as a tool to study the behavior of fibroblasts in cancer ( 34 ). The cells were introduced with an empty vector, ATF3 or ATF3 alternative isoform (ATF3ΔZip)—which lacks the leucine zipper DNA-binding domain ( 35 ) and therefore serves as a negative control for ATF3 transcriptional activity ( Figure 2A and Supplementary Figure S2 is available at Carcinogenesis Online). First, in a colony formation assay, all four fibroblastic cell lines overexpressing ATF3 displayed an increased capacity to form colonies when seeded at low cell density, which indicates that these cells gained a growth advantage under unfavorable conditions ( Figure 2B ). As mentioned above, ATF3 was elevated in the stromal compartment of clinical tumor samples ( Figure 1A ), which suggests that stromal ATF3 might promote tumor growth. To test this hypothesis, we injected lung fibroblasts overexpressing ATF3 (HK-3T ATF3 ) together with H-1299 human lung carcinoma cells into the hind limb of 10 nude mice. As a control, the parallel hind limb of each mouse was injected with HK-3T-expressing empty vector and H-1299 cells, generating a comparative setup, in which the capability of the fibroblasts to promote tumor growth can be measured. As depicted in Figure 2C , HK-3T ATF3 promoted the growth of the co-injected carcinoma cells, giving rise to larger tumors. Since H-1299 cells are highly tumorigenic and have an unstable genomic makeup ( 36 ), we wished to test whether stromal ATF3 can promote the growth of less aggressive tumor cells. To this end, we co-injected WI-38T cells expressing ATF3 (WI-38T ATF3 ) or an empty vector, with a lung fibrosarcoma-forming cell line (WI-38T Transformed ) that was generated previously in our laboratory ( 31 ). In agreement with the results obtained with HK-3T ATF3 cells, WI-38T ATF3 cells promoted the growth of the adjacent WI-38T Transformed cells ( Figure 2C ). Examination of the resulting tumor sections revealed malignant and rapidly growing tumors with a growth pattern that is cohesive and expansile ( Figure 2D ). Furthermore, as shown in Figure 2D and E , tumors that were co-injected with ATF3-expressing stroma had a significant enrichment in proliferating cells as can be inferred from the percentage of Ki67-positive cells. This result may account for their increased tumor size.
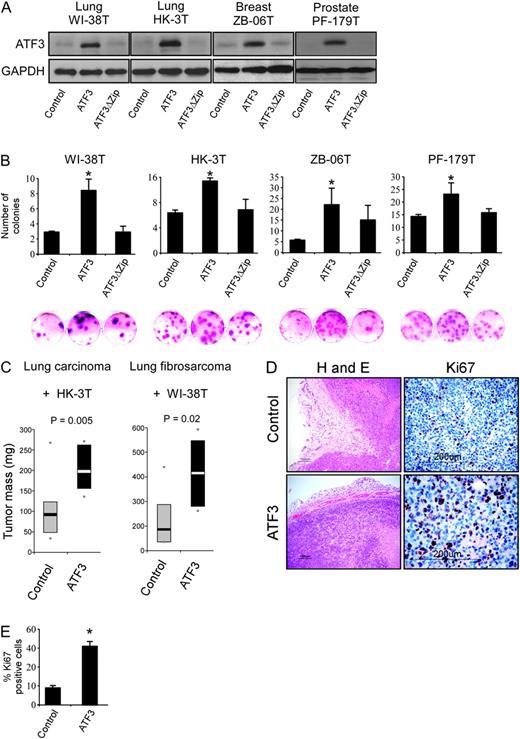
ATF3 promotes fibroblast colony-forming ability and tumor growth. ( A ) The designated fibroblast cultures were introduced with a retroviral empty vector or retroviruses encoding ATF3 or ATF3ΔZip. Western blot analysis was conducted for ATF3 and GAPDH as loading control. ( B ) Colony-forming efficiency assay. The designated cell cultures were seeded at a low cell density, and following 14 days, plates were stained with crystal violet. ‘*’Denotes a significant increase in colony formation in the ATF3 sample versus the empty control vector, P < 0.05. ( C ) (Left panel) HK-3T ATF3 or an empty vector were co-injected with H-1299 cells into 10 Nude mice. The bars represent 25–75% interval of tumor weight in milligrams, and the contrasted bars denote the median. Dots represent minimum and maximum weight in a given group. The same experiment was performed using WI-38T ATF3 and transformed WI-38T Transformed (right panel). ( D ) H-1299/HK-3T ATF3 -generated tumors were fixed, sectioned and stained with either hematoxylin and eosin or anti-Ki67. ( E ) Several 70 × 70 μm fields of view were scanned and the percentage of Ki67-positive cells were calculated, P = 0.008. For further information, see also Supplementary Figure S2 (available at Carcinogenesis Online).
Our results indicate that overexpression of ATF3 in CAFs confers them with a survival advantage and promotes the growth of adjacent tumor cells in a non-cell autonomous manner.
ATF3 facilitates a cancer-associated transcriptional program in fibroblasts
In an attempt to understand the molecular mechanism underlying ATF3 activity in the tumor–stroma interaction, we utilized microarrays to globally analyze mRNA expression of the two sets of the aforementioned lung stromal fibroblasts, namely HK-3T and WI-38T, expressing ATF3, ATF3ΔZip or an empty vector. As illustrated in Figure 3A , ATF3 expression in HK-3T lung fibroblasts resulted in upregulation of 80 genes and downregulation of 269 genes compared with the empty vector or ATF3ΔZip cells. In the WI-38T lung fibroblasts, ATF3 expression resulted in upregulation of 147 genes and downregulation of 172 genes. Notably, a large number of genes that were influenced by ATF3 expression (36 in the upregulated gene cluster and 131 in the downregulated gene cluster) overlapped between the two lung fibroblasts, as depicted in a Venn diagram ( Figure 3A , right panels), implying that the transcriptional regulation of these genes by ATF3 is not cell line specific and might represent a general transcription program facilitated by ATF3 in fibroblasts. A gene ontology clustering program [GeneDecks ( 37 )] revealed that these subsets of genes were significantly enriched with cancer-related clusters such as 'tumors', 'metastasis' and 'positive regulation of cellular proliferation', as well as with extracellular localization ( Supplementary Figure S3A is available at Carcinogenesis Online). Thus, ATF3 facilitates a general, cancer-related gene expression program in fibroblasts.
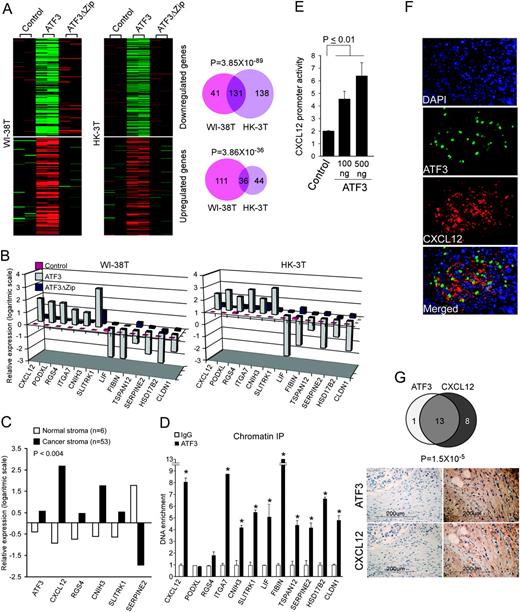
ATF3 facilitates a cancer-related transcriptional program in fibroblasts. ( A ) WI-38T and HK-3T were introduced with an empty vector or vectors encoding ATF3 or ATF3ΔZip and subjected to a complementary DNA microarray analysis. ATF3-induced/repressed gene clusters are shown. The numbers of the differentially expressed genes and the overlap between the two cell lines are shown in a Venn diagram (lower panels). Hypergeometric probabilities for the overlap between the gene lists are presented above the diagrams. ( B ) The expression level of 12 genes (six upregulated and six downregulated) was measured by quantitative real-time PCR and compared between WI-38T and HK-3T expressing ATF3, ATF3ΔZip or empty vector (control), P < 0.01. ( C ) A complementary DNA microarray analysis conducted by Finak et al . ( 32 ) comparing NAFs and CAFs was queried using the ‘Oncomine’ website ( 32 , 33 ) for the expression of the indicated genes. P -value for differential expression between NAFs and CAFs for all genes is smaller than 4 × 10 −3 . ( D ) A chromatin immunoprecipitation was done using an antibody against ATF3 or a control IgG antibody in HK-3T ATF3 cells. Quantitative real-time PCR was performed for precipitated DNA with primers that amplify regions surrounding the predicted ATF3/CREB-binding sites in the designated loci. Significant promoter enrichment was measured by two-tailed paired t -test. ‘*’Denotes P -value <0.02. ( E ) A vector encoding luciferase under the control of CXCL12 promoter and the indicated amounts of an expression vector encoding ATF3 were transfected into HT-1080 cells. The chart depicts normalized CXCL12 promoter activity calculated by dividing the luciferase activity in each well by the Renilla activity of co-transfected plasmid. ( F ) Tumors (WI-38 Transformed co-injected with WI-38T ATF3 ) were removed, embedded in paraffin and sectioned. Sections were stained with antibodies against CXCL12 or ATF3. Nuclei were visualized with 4′,6-diamidino-2-phenylindole (DAPI). ( G ) Breast cancer slides consisting of 47 samples were stained for ATF3 and CXCL12. (upper panel) A Venn diagram depicting the calculated overlap between ATF3 and CXCL12 stromal-positive samples. (lower panel) Representative images taken from matching samples stained for ATF3 and CXCL12, either negative (left hand) or positive (right hand). Supportive evidence is shown in Supplementary Figure S3 (available at Carcinogenesis Online).
To identify specific targets that might mediate the cancer-promoting activity of ATF3, we chose to focus on a group of 12 genes (six upregulated and six downregulated) that demonstrated a robust transcriptional response to the modulation of ATF3 in lung fibroblasts ( Figure 3B ), as well as in the two other fibroblastic systems, namely breast cancer-derived and prostate cancer-derived fibroblasts ( supplementary Figure S3B is available at Carcinogenesis Online). Importantly, in accordance with our in vitro analysis, five of the 12 genes were coregulated with ATF3 in a clinical study presented in the ‘Ocnomine’ database, comparing NAFs and breast CAFs ( 32 , 33 ) ( Figure 3C ). Furthermore, revisiting our previously published study comparing NAFs and CAFs, we found that ATF3, CLDN1, HSD17B2 and CXCL12 exhibited the same expression pattern in prostate CAFs ( 38 ) ( Supplementary Figure S3C is available at Carcinogenesis Online). A summary of the function of these genes and the number of their cancer-related publications is described in Table 1 . Interestingly, most of the genes (10 out of 12) were already investigated in the context of cancer, whereas the involvement of two genes, FIBIN and CNIH3 , in tumorigenesis is first proposed herein. In addition, this set of genes consists of secreted proteins (CXCL12, LIF, FIBIN and SERPINE2) and cell surface proteins (RGS4 and CLDN1) that can alter the tumor microenvironment scenery and may account for ATF3 non-cell autonomous mediated outcome. To investigate whether ATF3 effect on the expression of the 12 selected genes is mediated through DNA binding, we performed a Chromatin immunoprecipitation assay using HK-3T ATF3 . For the majority of the genes (10 out of 12), we detected binding of ATF3 to a predicted ATF3/CREB-binding sites (described in Supplementary Figure S3D and Supplementary Data is available at Carcinogenesis Online) in their promoters ( Figure 3D ). As CXCL12 (SDF-1a) is one of the key factors in the tumor–stroma interaction ( 40 ), we decided to perform an in-depth validation as for its regulation by ATF3, as a proof of concept. To this end, we cloned the promoter of CXCL12 into a luciferase reporter vector and co-transfected it into HT-1080 cells with an ATF3-expressing vector or an empty vector. Indeed, CXCL12 promoter was activated by ATF3 in a dose-dependent manner ( Figure 3E ). In addition, the aforementioned tumors, which were generated by co-injecting tumor and stromal cells, were sectioned and stained with antibodies against ATF3 and CXCL12. Strikingly, CXCL12 staining was evident exclusively in the Extra Cellular Matrix surrounding the ATF3-expressing cells ( Figure 3F ). As CXCL12 was reported previously to be elevated in breast cancer ( 41 ), we stained two consecutive slides consisting of 48 breast cancer specimens with α-ATF3 and α-CXCL12 antibodies. We found stromal expression of CXCL12 in 21 of the samples and 14 ATF3-positive stromal samples. More importantly, as depicted in Figure 3G , an overlap between the positive samples was evident in 13 cases (hypergeometric function, P = 1.5 × 10 −5 ), further substantiating the correlation between ATF3 and CXCL12 expression.
ATF3 target genes summary
| Gene symbol | Description/function a | Cancer-related references |
| ATF3 -upregulated genes | ||
| CXCL12 | A stromal cell-derived alpha chemokine, member of the intercrine family. | >30 |
| PODXL | Binds in a membrane protein complex with Na+/H+ exchanger, expressed in vascular endothelium cells. | 4 |
| RGS4 | GTPase-activating proteins for G alpha subunits of heterotrimeric G proteins. | 5 |
| ITGA7 | Belongs to the integrin alpha chain family receptor for the basement membrane protein laminin-1. | 5 |
| CNIH3 | Selective transport and maturation of TGFα family proteins. Upregulated in certain human mesenchymal stem cells. | 0 |
| SLITRK1 | Integral membrane proteins that are characterized by two N-terminal leucine-rich repeat domains. | 2 |
| ATF3 -downregulated genes | ||
| LIF | A pleiotropic cytokine involved in the induction of hematopoietic and neuronal differentiation. | >10 |
| FIBIN | A secreted lateral mesoderm signal protein essential for fin initiation in zebra fish. | 0 |
| TSPAN12 | A cell surface protein, belongs to the transmembrane 4 superfamily | 1 |
| SERPINE2 | A secreted Serpin peptidase inhibitor. | 7 |
| HSD17B2 | Hydroxysteroid (17-beta) dehydrogenase 2, responsible for the inactivation of estradiol. | 4 |
| CLDN1 | An integral membrane protein and a component of tight junction complexes | 16 |
| Gene symbol | Description/function a | Cancer-related references |
| ATF3 -upregulated genes | ||
| CXCL12 | A stromal cell-derived alpha chemokine, member of the intercrine family. | >30 |
| PODXL | Binds in a membrane protein complex with Na+/H+ exchanger, expressed in vascular endothelium cells. | 4 |
| RGS4 | GTPase-activating proteins for G alpha subunits of heterotrimeric G proteins. | 5 |
| ITGA7 | Belongs to the integrin alpha chain family receptor for the basement membrane protein laminin-1. | 5 |
| CNIH3 | Selective transport and maturation of TGFα family proteins. Upregulated in certain human mesenchymal stem cells. | 0 |
| SLITRK1 | Integral membrane proteins that are characterized by two N-terminal leucine-rich repeat domains. | 2 |
| ATF3 -downregulated genes | ||
| LIF | A pleiotropic cytokine involved in the induction of hematopoietic and neuronal differentiation. | >10 |
| FIBIN | A secreted lateral mesoderm signal protein essential for fin initiation in zebra fish. | 0 |
| TSPAN12 | A cell surface protein, belongs to the transmembrane 4 superfamily | 1 |
| SERPINE2 | A secreted Serpin peptidase inhibitor. | 7 |
| HSD17B2 | Hydroxysteroid (17-beta) dehydrogenase 2, responsible for the inactivation of estradiol. | 4 |
| CLDN1 | An integral membrane protein and a component of tight junction complexes | 16 |
Gene descriptions/functions were taken from National Center for Biotechnology Information ‘Gene’ tool ( 39 ).
ATF3 target genes summary
| Gene symbol | Description/function a | Cancer-related references |
| ATF3 -upregulated genes | ||
| CXCL12 | A stromal cell-derived alpha chemokine, member of the intercrine family. | >30 |
| PODXL | Binds in a membrane protein complex with Na+/H+ exchanger, expressed in vascular endothelium cells. | 4 |
| RGS4 | GTPase-activating proteins for G alpha subunits of heterotrimeric G proteins. | 5 |
| ITGA7 | Belongs to the integrin alpha chain family receptor for the basement membrane protein laminin-1. | 5 |
| CNIH3 | Selective transport and maturation of TGFα family proteins. Upregulated in certain human mesenchymal stem cells. | 0 |
| SLITRK1 | Integral membrane proteins that are characterized by two N-terminal leucine-rich repeat domains. | 2 |
| ATF3 -downregulated genes | ||
| LIF | A pleiotropic cytokine involved in the induction of hematopoietic and neuronal differentiation. | >10 |
| FIBIN | A secreted lateral mesoderm signal protein essential for fin initiation in zebra fish. | 0 |
| TSPAN12 | A cell surface protein, belongs to the transmembrane 4 superfamily | 1 |
| SERPINE2 | A secreted Serpin peptidase inhibitor. | 7 |
| HSD17B2 | Hydroxysteroid (17-beta) dehydrogenase 2, responsible for the inactivation of estradiol. | 4 |
| CLDN1 | An integral membrane protein and a component of tight junction complexes | 16 |
| Gene symbol | Description/function a | Cancer-related references |
| ATF3 -upregulated genes | ||
| CXCL12 | A stromal cell-derived alpha chemokine, member of the intercrine family. | >30 |
| PODXL | Binds in a membrane protein complex with Na+/H+ exchanger, expressed in vascular endothelium cells. | 4 |
| RGS4 | GTPase-activating proteins for G alpha subunits of heterotrimeric G proteins. | 5 |
| ITGA7 | Belongs to the integrin alpha chain family receptor for the basement membrane protein laminin-1. | 5 |
| CNIH3 | Selective transport and maturation of TGFα family proteins. Upregulated in certain human mesenchymal stem cells. | 0 |
| SLITRK1 | Integral membrane proteins that are characterized by two N-terminal leucine-rich repeat domains. | 2 |
| ATF3 -downregulated genes | ||
| LIF | A pleiotropic cytokine involved in the induction of hematopoietic and neuronal differentiation. | >10 |
| FIBIN | A secreted lateral mesoderm signal protein essential for fin initiation in zebra fish. | 0 |
| TSPAN12 | A cell surface protein, belongs to the transmembrane 4 superfamily | 1 |
| SERPINE2 | A secreted Serpin peptidase inhibitor. | 7 |
| HSD17B2 | Hydroxysteroid (17-beta) dehydrogenase 2, responsible for the inactivation of estradiol. | 4 |
| CLDN1 | An integral membrane protein and a component of tight junction complexes | 16 |
Gene descriptions/functions were taken from National Center for Biotechnology Information ‘Gene’ tool ( 39 ).
Stromal ATF3 promotes cancer progression through the regulation of CXCL12, RGS4 and CLDN1
To determine the identity of the most prominent mediators of ATF3 tumor-promoting activity, we reversed the expression of ATF3-regulated genes by either overexpression or short hairpin-mediated knockdown in HK-3T ATF3 . These manipulations were performed on the background of high ATF3 levels, in an attempt to reverse ATF3 tumor-promoting capacity. Figure 4A illustrates the mRNA levels of the genes, following stable infections. Due to the fact that we could not observe any reduction in CNIH3 mRNA following infection of four different short hairpin RNA constructs, we decided to exclude it from the experiment. First, we tested whether reversing the transcriptional levels of ATF3 target genes will affect the ability of the stromal cells to form colony in unfavorable condition (as shown in Figure 2B ). Indeed, reversing the transcriptional levels of the ATF3 targets LIF, CLDN1, SERPINE2, HSD17B2 PODXL and ITGA7 attenuated the colony-forming capacity of HK-3T ATF3 cells ( Figure 4B ), suggesting that these genes mediate the growth-facilitating effect of ATF3 in the stroma. More importantly, utilizing the aforementioned co-injection experimental setup ( Figure 2C ), we found that CAFs overexpressing CLDN1 and CAFs with a knocked down expression of RGS4 and CXCL12 profoundly reduced the adjacent tumor size ( Figure 4C ). Reversal of the remaining genes did not have a significant effect on tumor size (data not shown). These results indicate that by repressing CLDN1 expression and by transactivating CXCL12 and RGS4 , stromal ATF3 exerts a tumor-promoting activity.
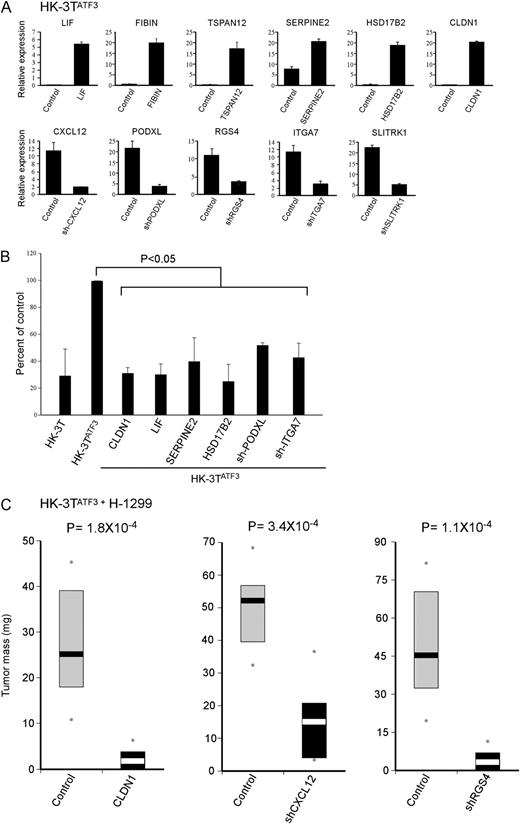
ATF3 effect on tumor growth is mediated via a subset of genes. HK-3T ATF3 was introduced either with a vector encoding an ATF3-downregulated target or with a vector expressing a short hairpin against an ATF3-upregulated target. Corresponding empty vectors were used as controls for each of the cases. ( A ) Quantitative real-time PCR analysis was performed to measure the designated genes’ expression, P < 0.05 ( B ) Colony-forming efficiency assay. The designated cell cultures were seeded at a low cell density and following 14 days plates were stained with crystal violet. ( C ) HK-3T ATF3 cells were co-injected with H-1299 cells into 10 Nude mice. Bars represent 25–75% interval of tumor weight in milligrams, and the contrasted bars denote the median. Dots represent minimum and maximum weight in a given group.
Discussion
Although ATF3 has been thoroughly investigated in tumor cells, up to date, the possible role of ATF3 in the tumor stroma has not been reported. In our current work, we chose to pursue the possible influence of ATF3 in the stromal microenvironment. We demonstrate here for the first time that ATF3 is frequently expressed in the stroma of several cancers, which extends the spectrum of ATF3 activity to the vicinity of the tumor and raises new possibilities as for its involvement in the process of tumorigenesis. Our findings invoke several intriguing questions that are addressed in this discussion; among them are ATF3 deregulation in cancer, the identity and consistency of its targets and the alluring potential of targeting stromal ATF3, as a mean to control cancer progression.
ATF3 deregulation in cancer
The ample data regarding ATF3 portray it as a dichotomous protein, which may act as a tumor suppressor, protecting normal and cancer cells from further transformation, but can also facilitate tumor progression ( 15 , 25 ). Several mechanisms may account for ATF3 dichotomy; among them are its different heterodimerization partners, which may lead to the activation or repression of different target genes. In addition, as a transcription factor, it might be greatly affected by a given chromatin availability state, which can be tumor stage or type dependent. As a stress response protein, all normal cells benefit from ATF3 upregulation in response to various insults. However, its constitutive expression in a large portion of cancers suggests that in the long run, ATF3 may exert oncogenic properties, which are positively selected by the tumor cells, and according to our present findings, in their surrounding stroma, as well. Indeed, ATF3 was reported to be overexpressed in cancer due to increased genomic copy number ( 24 , 25 ). The transition between the tumor-suppressive and oncogenic state could be attributed to posttranslational modifications. Notably, ATF3 amino acid sequence is enriched with potential phosphorylation, acetylation, methylation, ubiquitination and sumoylation sites, spanning 20% of the protein ( 42 ). Future experimental studies should be aimed at understanding whether ATF3 activity is mediated via posttranslational modifications—variations which may account for its dichotomy in cancer both in the tumor and its surrounding cells.
The identity of ATF3′s targets
Two principal selective engines shape the behavior and gene expression of tumor-associated stromal cells. The first is increased survival and enhanced growth rate often exhibited by CAFs ( 38 , 43 ), which grant the cells with a growth advantage over other inhabitants of the tumor stroma milieu. A second mechanism is the selective force imposed by the developing tumor, which favors only the stromal cells that can supply its growing demands. For example, in a seminal work by Hill et al. ( 43 ), tumor cells were shown to actively select for highly prolific fibroblasts, which harbor p53 mutations. As depicted in Figure 5 , our findings suggest that the identity of ATF3 targets in the stromal cells is governed by these two selective forces. On the one hand, LIF, CLDN1, SERPINE2, HSD17B2, ITGA7 and PODXL mediate the increased colony-forming ability of the fibroblasts following ATF3 expression. In contrast, despite not affecting the immediate growth of stromal cells, CXCL12 and RGS4 induction by ATF3 enhanced tumor growth in a non-cell autonomous manner. Moreover, these genes were also found to be co-expressed with ATF3 in cancer-associated stromal cells that were isolated from clinical samples. Thus, tumor selection may account for their ATF3-mediated upregulation. ATF3 seems to activate a plethora of genes and pathways all funneled into the regulation of cancer progression. This could stem from the fact that most of these genes (10 out of 12) were shown to exert a substantial effect (either suppression or promotion) on tumor progression independently of ATF3. Thus, reversing their expression had a noticeable impact on both the colony-forming capacity and tumor-promoting phenotype.
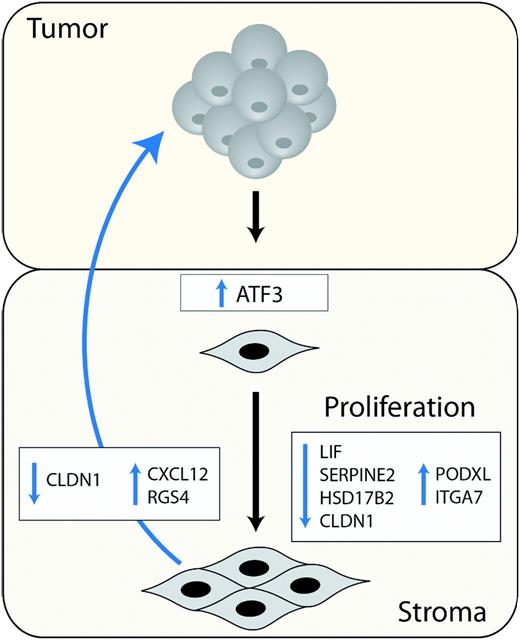
A schematic model summarizing stromal ATF3 activity in cancer.
ATF3 as a potential target for anticancer therapy
In contrast to a wide range of transcriptionally activated and repressed targets of ATF3 in different tumors ( 15 ), which renders ATF3 transcriptional activity as ‘cell-type specific’, the identity of ATF3 targets in various stromal cells seems to be consistent. This incongruity might stem from the frequent genomic instability observed in cancer compared with the relatively stable genomes of stromal cells ( 7 , 44 ). Since stromal ATF3 overexpression occurs in a relatively stable genomic background and since it executes a general transcriptional program in fibroblasts, which is not dependent on the specific tumor, it might be a suitable candidate for stromal-targeted therapy in multiple cancers. Moreover, CXCL12, which was reported previously to be expressed in many cancer-related stromal cells ( 32 , 38 ), is described here as a novel target of ATF3. CXCL12 is a cancer-related chemokine that binds CXCR4 and CXCR7 receptors ( 45–48 ) and regulates cell survival, proliferation and chemotaxis ( 49 ). More importantly, Orimo et al. ( 40 ), reported that CAFs promoted tumor growth through secretion of CXCL12, which on the one hand induce angiogenesis and on the other hand directly enhance tumor growth by binding and activating CXCR4 receptor on the surface of tumor cells. Accordingly, CXCL12 and its interaction with downstream components are currently being targeted in stromal-related cancer therapy, and two such drugs (AMD3100 and CTCE-9908) had been approved for clinical use ( 47 , 50 ). As ATF3 was shown to promote endothelial survival and sprouting ( 17 ) and as it is a regulator of CXCL12, it is tantalizing to speculate that ATF3 inhibition will result in attenuation of tumor growth as a result of insufficient angiogenesis.
Supplementary material
Supplementary Table S1 and Supplementary Data – Supplementary Data can be found at http://carcin.oxfordjournals.org/ .
Funding
Center of Excellence grant from Flight Attendant Medical Research Institute, Yad Abraham Center for Cancer Diagnosis and Therapy and by European Community FP7-INFLACARE number-223151.
Abbreviations
- ATF3
activating transcription factor 3
- CAF
cancer-associated fibroblast
- mRNA
messenger RNA
- NAF
normal-associated fibroblast
- PCR
polymerase chain reaction
S.M. wishes to thank Dr Eudice Goldberg once again for her kind support. This publication reflects the authors' views and not necessarily those of the European Community. The European Community is not liable for any use that may be made of the information contained herein. V.R. is the incumbent of the Norman and Helen Asher Professorial Chair Cancer Research at the Weizmann Institute.
Conflict of Interest Statement: None declared.
References
Author notes
These authors contributed equally to this work

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}