




Abstract
Pancreatic ductal adenocarcinoma (PDAC) is a leading cause of cancer mortality in Western countries. We have shown previously that four representative human PDAC cell lines were regulated by beta-adrenoreceptors via cyclic adenosine 3′,5′-monophosphate (cAMP)-dependent signaling. In the current study, we have tested the hypothesis that nicotine stimulates the growth of PDAC xenografts in nude mice by increasing the systemic levels of the stress neurotransmitters adrenaline and noradrenaline, which are the physiological agonists for beta-adrenoreceptors and that inhibition by γ-aminobutyric acid (GABA) of the adenylyl cyclase-dependent pathway downstream of adrenoreceptors blocks this effect. The size of xenografts from PDAC cell line Panc-1 was determined 30 days after inoculation of the cancer cells. Stress neurotransmitters in serum as well as cAMP in the cellular fraction of blood and in tumor tissue were assessed by immunoassays. Levels of GABA, its synthesizing enzymes GAD65 and GAD67 and beta-adrenergic signaling proteins in the tumor tissue were determined by western blotting. Nicotine significantly increased the systemic levels of adrenaline, noradrenaline and cAMP while increasing xenograft size and protein levels of cAMP, cyclic AMP response element-binding protein and p-extracellular signal-regulated kinase 1/2 in the tumor tissue. Nicotine additionally reduced the protein levels of both GAD isozymes and GABA in tumor tissue. Treatment with GABA abolished these responses to nicotine and blocked the development of xenografts in mice not exposed to nicotine. These findings suggest that the development and progression of PDAC is subject to significant modulation by stimulatory stress neurotransmitters and inhibitory GABA and that treatment with GABA may be useful for marker-guided cancer intervention of PDAC.
Introduction
Pancreatic cancer is the fourth leading cause of cancer mortality in both men and women in Western countries ( 1 ) and smoking, diabetes and pancreatitis are risk factors ( 2 ). More than 95% of all pancreatic cancers are pancreatic ductal adenocarcinomas (PDACs). PDAC is one of the most aggressive cancers with extensive invasiveness and metastasis precluding surgical resection in the majority of patients at the time of diagnosis. In addition, PDAC is unresponsive to conventional radio- and chemotherapy, resulting in a mortality rate near 100% within 1 year of diagnosis ( 3 ). Molecular targeting of therapy for PDAC currently emphasizes the epidermal growth factor receptor pathway that is frequently overexpressed in PDAC ( 4 ). While this strategy prolongs the survival of patients by several months when started after resection of operable PDACs, the majority of patients present with metastatic or advanced local disease and fail to respond to these agents. There is thus an urgent need for novel strategies that prevent the development of PDAC in individuals at risk.
The stress neurotransmitters adrenaline and noradrenaline are catecholamines that are synthesized and released into the systemic circulation by neurons in response to nicotinic acetylcholine receptor (nAChR) stimulation in the central and peripheral nervous system and in the adrenal medulla ( 5 , 6 ). Both catecholamines regulate diverse organ and cell functions by binding as agonists to alpha- and beta-adrenoreceptors, which are members of the G-protein-coupled receptor family. Binding of an agonist to these receptors activates the stimulatory G-protein Gα s . In turn, Gα s activates adenylyl cyclase, the rate-limiting enzyme for the formation of intracellular cyclic adenosine 3′,5′-monophosphate (cAMP). Nicotine is a selective agonist for nAChRs ( 7 ) and stimulates the synthesis and release of adrenaline and noradrenaline by binding to these receptors ( 8–10 ). The resulting increase in the systemic levels of these stress neurotransmitters is an important factor in the development of smoking-associated cardiovascular disease ( 11 ).
Using four representative human PDAC cell lines and immortalized human pancreatic duct epithelial cells, we have shown that the proliferation, migration and apoptosis of these cells are regulated by beta-adrenoreceptors via cAMP-dependent signaling, resulting in the activation of protein kinase A (PKA)/cyclic AMP response element-binding protein (CREB), transactivation of the epidermal growth factor receptor and activation of caspase-3 ( 12–14 ). In addition, these studies showed that the powerful tobacco carcinogen 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone (NNK) is a high-affinity agonist for beta-adrenoreceptors that stimulates PDAC cells via activation of these signaling pathways. While these findings provided a first direct link between a tobacco-specific carcinogen and the regulation of PDAC, they also suggested that a systemic increase in adrenaline/noradrenaline might contribute to the development and progression of PDAC.
The amino acid neurotransmitter γ-aminobutyric acid (GABA) is the major inhibitory neurotransmitter in the central nervous system that counteracts the stimulatory catecholamine neurotransmitters by binding to the GABA B receptor that inhibits adenylyl cyclase via Gα i ( 15 ). Recent studies have shown that GABA was suppressed in 29 of 30 investigated human PDACs, whereas noradrenaline, PKA, p-CREB and p-extracellular signal-regulated kinase (ERK) were overexpressed ( 14 ). In addition, GABA significantly inhibited base level and beta-adrenoreceptor-stimulated PDAC cell growth and migration in vitro , responses blocked by genetic silencing of the GABA B receptor ( 14 ). These findings suggest that GABA may have tumor suppressor function for PDAC. Beta cells of the pancreatic islets are an important source of GABA in the pancreas ( 15 ). The number of these cells is greatly reduced in people with diabetes and pancreatitis. In addition, smoking decreases GABA levels by as yet unidentified mechanisms ( 16 ). The resulting deficit in inhibitory GABA signaling may create an environment favorable for the development of PDAC in smokers with pancreatitis or diabetes.
Prompted by first reports that nicotine modulates intracellular signaling pathways that regulate cell proliferation and apoptosis in small cell lung cancer cells ( 17–20 ), nicotinic receptor-mediated pro-mitotic and anti-apoptotic intracellular signaling has been described in numerous other cancers and cells ( 21 , 22 ). While these studies have focused on direct intracellular signaling downstream of nAChRs in cancer cells, the central regulatory role of nAChRs in the systemic levels of stimulatory and inhibitory neurotransmitters has been given little attention by cancer researchers. In the current study, we have tested the hypothesis that nicotine stimulates the growth of PDAC xenografts in nude mice by increasing the systemic levels of adrenaline and noradrenaline and that inhibition by GABA of the adenylyl cyclase-dependent beta-adrenergic pathway stimulated by these agents blocks this effect.
Materials and methods
Animal experiment
The animal experiment was approved by the Institutional Animal Care and Use Committee. Male athymic nude mice (Harlan Sprague Dawley, Indianapolis, IN) were housed in our Experimental Animal Facility (10 mice per cage) under standard laboratory conditions with free access to food (Purina Rodent Chow) and water. Each mouse was inoculated in the flank region by subcutaneous injection with cells (3 × 10 6 , viability >95%) suspended in 0.2 ml of phosphate-buffered saline of the human PDAC cell line Panc-1 (American Type Culture Collection, Manassas, VA). The mice were then randomly assigned to the following treatment groups (10 mice per group):
Group 1: Sterile tap water control.
Group 2: 200 μg/ml nicotine (Sigma, St Louis, MO) in sterile drinking water.
Group 3: Sterile tap water + GABA (Sigma; 10 mg/kg body weight) by intraperitoneal injection 5 days/week.
Group 4: 200 μg/ml of nicotine in sterile drinking water + GABA (10 mg/kg body weight) by intraperitoneal injection 5 days a week.
The dose of nicotine used is within the range of daily nicotine intake in heavy smokers ( 23 ). Treatment with nicotine and GABA started 1 day after subcutaneous implantation of the tumor cells. All animals were observed for 30 days while being monitored daily by a staff veterinarian board certified in Laboratory Animal Science. Body weights were recorded once a week. Liquid consumption was monitored by measuring the tap water with and without the nicotine in and out every day. At the end of the 30-day observation period, the animals were euthanized by CO 2 inhalation. Xenograft volumes were calculated from two perpendicular diameters of each tumor measured using the formula: volume = (length/2) × (width 2 ). Mean values and standard deviations were calculated and differences among groups tested by one-way analysis of variance (ANOVA) and Tukey–Kramer multiple comparison test. Blood samples were collected by heart puncture for the determination of adrenaline and noradrenaline in the serum and of cAMP in the cellular fraction of blood. The tumors were excised and snap frozen in liquid nitrogen for analysis by immunoassay and western blotting.
Analysis of serum adrenaline and noradrenaline
Quantitative analysis of systemic levels of adrenaline and noradrenaline in serum samples was conducted with an enzyme immunoassay according to the manufacturer's instruction (2-CAT enzyme-linked immunosorbent assay, Rocky Mountain Diagnostic, Colorado Spring, CO). Absorbance was read with an enzyme-linked immunosorbent assay reader at 450 nm. The quantification of the serum samples was achieved by comparing their absorbance with a reference curve prepared with known standard concentrations of adrenaline and noradrenaline. Mean values and standard errors from triplicate samples per treatment group were calculated. Statistical significance of data was assessed by one-way ANOVA, Tukey–Kramer multiple comparisons test and two-tailed unpaired t -test.
Analysis of cAMP in blood cells and tumor tissues
The cellular fraction of blood samples comprising erythrocytes, lymphocytes, granulocytes and thrombocytes was used for the analysis of systemic cAMP. Each of these blood cells expresses beta-adrenoreceptors, and lymphocytes have previously been used in human patients for the assessment of systemic cAMP-dependent signaling activity ( 24 ). After three washes with water, cells were treated with 0.1 M HCL for 20 min and then lysed by sonication. The levels of cAMP in tumor tissues and in skin tissue of the GABA-treated group without tumors were also determined after lysis of the tissue samples using a direct cAMP enzyme immunoassay kit according to the manufacturer's instructions (Assay Designs, Ann Arbor, MI). Color intensity was measured at 405 nm. Data were expressed as mean values and standard errors of triplicate samples per treatment group. Statistical analysis of data was by one-way ANOVA, Tukey–Kramer multiple comparisons test and two-tailed unpaired t -test.
Analysis of signaling proteins by western blotting
Following lysis of the thawed tissues, protein was determined using the BCA Protein Assay (Pierce, Rockford, IL). Equal amounts of protein were treated with loading buffer at 100°C for 5 min and applied to each lane for electrophoresis in 12% polyacrylamide gel. The electrophoresed proteins were transferred onto nitrocellulose membranes in transfer buffer at 100 mV for 1 h, treated with blocking buffer (5% non-fat dry milk in tris-buffered saline with Tween 20) for 1 h and incubated with primary antibody overnight at 4°C. The following primary antibodies were used: total CREB (Upstate Biotechnology, Lake Placid, NY); p-CREB, p-ERK1/2 and ERK1/2 (Cell Signaling, Danvers, MA); anti-GAD65, anti-GAD67 and anti-GABA (Millipore, Billerica, MA) and anti-beta-actin (Sigma). Following incubation with horseradish peroxidase-labeled secondary antibody (goat anti-mouse or goat anti-rabbit, Cell Signaling) for 1 h, immunoreactive bands were detected by chemiluminescent reaction (Enhanced chemiluminescence, Amersham Biosciences, Piscataway, NJ) via autoradiography on Kodak Bio-Max XAR film. Three separate western blots were conducted for each antibody per sample and yielded similar data. Relative densities of the bands were determined by image analysis using NIH SCION image analysis software. Mean values and standard errors from five densitometric readings per band were analyzed by one-way ANOVA and Tukey–Kramer multiple comparison test.
Results
All mice survived until the end of the 30-day observation period after inoculation with Panc-1 cells. Body weights of untreated mice and mice treated with nicotine alone or nicotine plus GABA were not statistically different. The animals treated with GABA alone had a 10% higher body weight than animals in the three other groups. None of the animals demonstrated abnormalities in behavior. The daily intake of water per mouse did not differ significantly among treatment groups. The animals receiving nicotine treatments consumed 6.9 ± 0.5 ml of water per day resulting in a daily dose of 1.4 ± 0.4 mg nicotine per mouse. Palpable xenografts were not detected in any of the mice treated with GABA alone prior to the end of the study. At necropsy, two of the mice treated with nicotine plus GABA showed microscopically detectable xenografts 0.18 and 0.17 cm 3 in volume, whereas microscopic investigation detected no tumors in mice treated with GABA alone. In the untreated mice and in the animals treated with nicotine alone, eight mice per each group showed palpable tumor xenografts 2 weeks after cancer cell inoculation. However, the volume of xenografts in untreated mice remained small until the end of the 30-day observation period (0.9 ± 0.2 cm 3 ), whereas xenograft volumes had increased 4.2-fold ( P < 0.001) in the animals given nicotine alone ( Figure 1 ). These data suggest a strong tumor-promoting effect of nicotine and strong antitumorigenic effects of GABA in untreated and nicotine-treated mice.
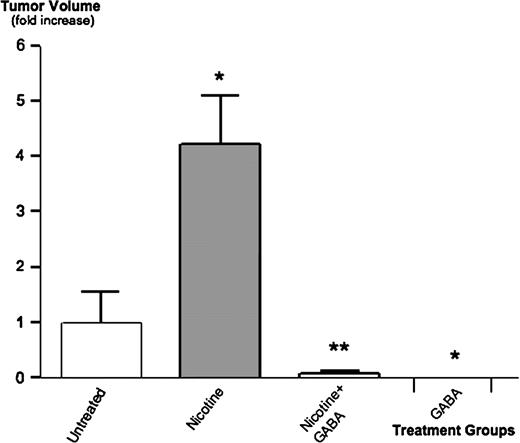
Effects of nicotine and GABA on Panc-1 xenografts. Athymic nude mice were subcutaneously injected with human PDAC cells Panc-1 and observed for 30 days. Controls received no pharmacological treatment, one group was given nicotine in the drinking water for 30 days, one group received nicotine in the drinking water and additional intraperitoneal injections with GABA (five times per week) and one group was treated by intraperitoneal injections with GABA alone. At the end of the observation period, nicotine had significantly ( P < 0.001) increased xenograft size, a response significantly ( P < 0.001) inhibited by GABA. Data were mean values and standard errors (expressed as fold increase over untreated animals) of the eight tumor-bearing mice in the untreated and nicotine-treated groups and of the two mice that developed small xenografts in the group treated with nicotine plus GABA. None of the animals given GABA alone developed xenografts. * significantly ( P < 0.001) different from untreated group; ** significantly ( P < 0.001) different from group treated with nicotine alone.
Immunoassays conducted with the serum fractions of blood samples showed base levels of adrenaline and noradrenaline in untreated animals of 12.1 ± 1.9 and 74.4 ± 2.5 ng/ml, respectively. Treatment with nicotine alone increased the levels of noradrenaline 4.2-fold ( P < 0.001) and of adrenaline 2-fold ( P < 0.001; Figure 2A ). Both responses were reduced to control levels ( P < 0.001) by the simultaneous treatment with GABA, whereas animals given GABA alone had catecholamine levels slightly below that of controls ( Figure 2 ). Measurement of systemic cAMP levels by immunoassay in the cellular fraction of blood samples revealed a significant ( P < 0.001) increase (1.86-fold) in the mice treated with nicotine alone ( Figure 2B ). This response was completely abolished by treatment with GABA, whereas treatment with GABA alone reduced cAMP levels to 0.6-fold of controls ( P < 0.001, Figure 2B ). In accord with these findings, cAMP was also significantly ( P < 0.001) increased (2.6-fold) in the tumor tissues of mice treated with nicotine alone and this response was blocked by GABA ( Figure 2B ). Taken together, these data indicate that nicotine caused an increase in intracellular cAMP in the systemic circulation and in the tumor tissues that were associated with elevated systemic levels of the catecholamine stress neurotransmitters. GABA abolished these effects and additionally reduced systemic catecholamine and cAMP levels below those in control animals.
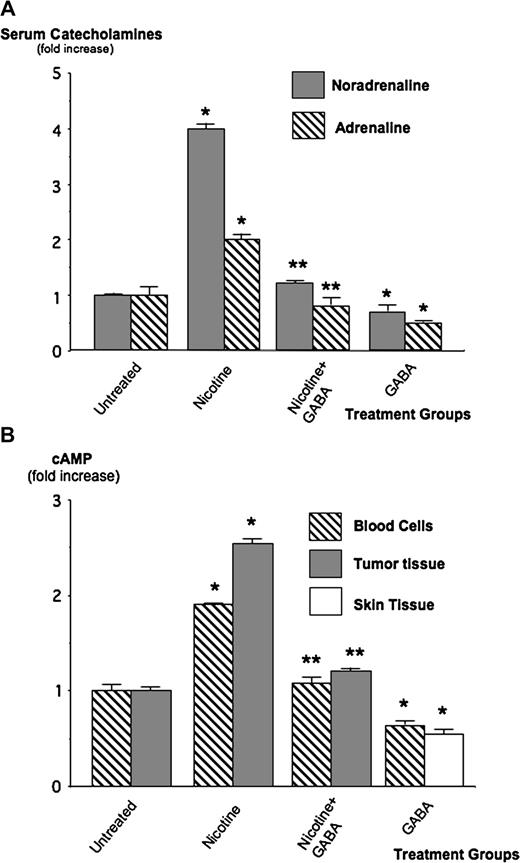
A ) Modulation of serum levels of adrenaline and noradrenaline by nicotine and GABA. Immunoassay revealed a 4.2-fold increase of noradrenaline ( P < 0.001) and a 2-fold increase of adrenaline ( P < 0.001) in the mice treated with nicotine. GABA completely abrogated these responses to nicotine. Data were mean values and standard errors of triplicate samples expressed as fold increase over the levels in untreated mice. * significantly ( P < 0.001) different from untreated group; ** significantly ( P < 0.001) different from group treated with nicotine alone. ( B ) Results of immunoassay for the determination of intracellular cAMP in the cellular fraction of blood samples in tumor tissue and in the skin of mice without tumors after treatment with GABA alone. Treatment with nicotine increased intracellular cAMP 1.86-fold in blood cells and 2.6-fold in xenograft tissue, a response completely blocked by GABA. Treatment with GABA alone significantly ( P < 0.001) reduced cAMP levels below the base levels in untreated animals. Data were mean values and standard errors of triplicate samples per treatment group expressed as fold increase over the levels in untreated animals. * significantly ( P < 0.001) different from untreated group; ** significantly ( P < 0.001) different from group treated with nicotine alone.
Analysis of protein expression by western blots from lysed tumor tissue showed that nicotine treatment significantly suppressed the expression of the GABA-synthesizing enzyme GAD65 ( P < 0.001) as well as GABA ( P < 0.001; Figure 3 ). In contrast, nicotine did not significantly affect the expression levels of GAD67 that was only present at low levels in the xenograft tissues from untreated animals ( Figure 3 ). Treatment of the nicotine-exposed mice with GABA significantly ( P < 0.001) inhibited the suppression of GAD65 and GABA while increasing GAD67 levels above that observed in untreated mice ( Figure 3 ). The skin samples taken from the mice in the group treated with GABA alone (none of which developed detectable xenograft tumors) demonstrated significantly ( P < 0.001) higher levels of GAD67 and GABA than tumor tissues from the untreated control group ( Figure 3 ). Collectively, these findings suggest that nicotine strongly inhibited the GABA system in PDAC xenografts, primarily via suppression of GAD65, and that treatment with GABA reversed these effects.
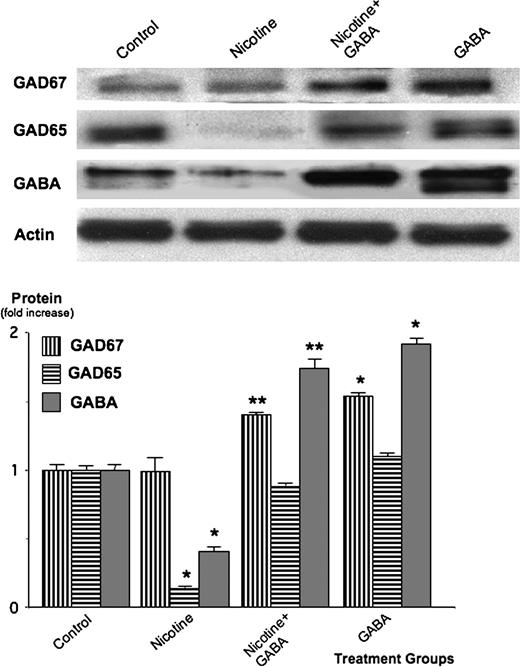
Western blots showing protein expressions of the GABA-synthesizing enzymes GAD67, GAD65 and the inhibitory neurotransmitter GABA in xenografts of untreated mice (controls), animals treated with nicotine or with nicotine plus GABA and in skin samples from the group treated with GABA alone. Nicotine significantly ( P < 0.001) suppressed GAD65 and GABA, an effect inhibited ( P < 0.001) by treatment with GABA. The columns in the graph are mean values and standard errors of five densitometric readings per band calculated as ratio of GAD or GABA protein over actin and expressed as fold increase over controls. * significantly ( P < 0.001) different from untreated group; ** significantly ( P < 0.001) different from group treated with nicotine alone.
We have shown previously in human PDAC cells Panc-1 and BXPC-3 in vitro that beta-adrenergic stimulatory signaling activates the transcription factor CREB, while additionally activating the mitogen-activated kinases ERK1/2 via PKA-mediated transactivation of the epidermal growth factor receptor (EGFR) ( 13 , 25 ). We therefore assessed the protein expressions of p-CREB and p-ERK1/2 by western blots to determine their modulation by nicotine and GABA in panc-1 xenografts. As Figure 4 shows, nicotine significantly ( P < 0.001) induced p-CREB (2.8-fold) and p-ERK1/2 (2.4-fold). The nicotine-induced increase in p-CREB was completely abrogated ( P < 0.001) when the animals were treated simultaneously with GABA, whereas p-ERK1/2 was significantly ( P < 0.001) but not completely reduced to control levels. The normal skin samples of the non-tumor-bearing mice in the group treated with GABA alone showed significantly ( P < 0.001) lower levels of p-CREB and p-ERK1/2 than the tumor tissues from the untreated controls ( Figure 4 ). Taken together, these findings indicate that nicotine strongly increased tumor-stimulating signaling via p-CREB and p-ERK, whereas GABA significantly inhibited this signaling cascade.
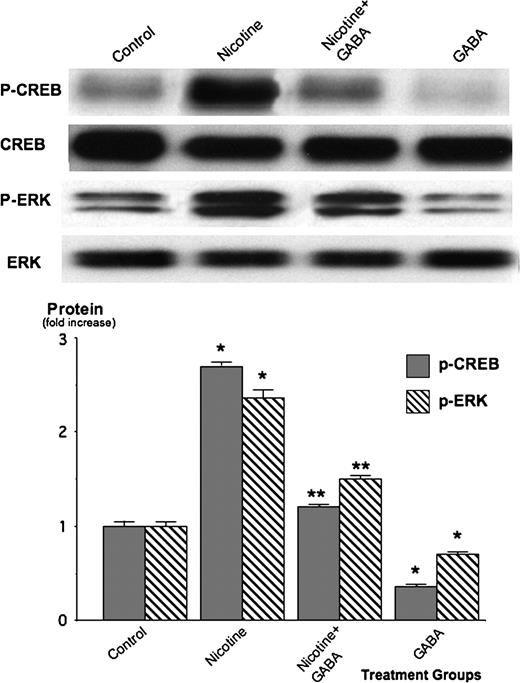
Western blots showing protein expression of phosphorylated and unphosphorylated CREB as well as phosphorylated and unphosphorylated ERK1/2 in xenografts of untreated mice (controls), animals treated with nicotine or with nicotine plus GABA and in skin samples from the group treated with GABA alone. Nicotine significantly ( P < 0.001) induced the expression levels of p-CREB and p-ERK1/2. These effects were inhibited ( P < 0.001) by GABA. The columns in the graph are mean values and standard errors of five densitometric readings per band calculated as ratio of GAD or GABA protein over actin and expressed as fold increase over controls. * significantly ( P < 0.001) different from untreated group; ** significantly ( P < 0.001) different from group treated with nicotine alone.
Discussion
Our data provide evidence, for the first time, that nicotine contributes to the development of smoking-associated PDAC by increasing the systemic levels of the stress neurotransmitters adrenaline and noradrenaline. The observed significant increase in systemic and tumor cAMP in conjunction with a significant induction of p-CREB and p-ERK1/2 in tumor cells of animals with elevated stress neurotransmitter levels supports the importance of cAMP signaling for this tumor-promoting effect that was first suggested by in vitro studies ( 12–14 ) in four representative human PDAC cell lines (including Panc-1) and in normal human duct epithelial cells. While in the current experiment the increase in stress neurotransmitters was caused by treatment of the mice with nicotine, stress, anxiety and numerous lifestyle factors can have similar effects. In addition, many foods, beverages, dietary supplements and weight loss regimens that increase energy either stimulate beta-adrenoreceptors or increase their responsiveness by phosphodiesterase inhibitors that block cAMP breakdown. Patients undergoing treatment for PDAC would thus probably benefit from a careful analysis and adjustment of psychological, dietary and lifestyle factors to reduce potentially hyperactive cAMP signaling. This adjuvant strategy may also benefit patients with several other types of cancer. It has thus been shown that epinephrine and norepinephrine enhance the metastasis and progression of ovarian cancer ( 27 ). In addition, human cancer cell lines derived from adenocarcinomas of the lungs ( 28 , 29 ), breast ( 30 ), stomach ( 31 ), colon ( 32 ), and prostate ( 33 ) are stimulated in their proliferation and/or migration by beta-adrenergic agonists.
The observed strong inhibition of basal and nicotine-induced xenograft growth and cAMP signaling by GABA represent the first in vivo evidence for the potential usefulness of this neurotransmitter in the prevention and treatment of PDAC. Suppressed GABA levels accompanied by overexpressed noradrenaline, PKA, p-CREB and p-ERK were recently reported by us in 29 of 30 investigated tissue arrays from human PDACs ( 14 ). In addition, we showed that GABA inhibited cAMP signaling, cAMP-dependent transactivation of the EGFR pathway, cell proliferation and cell migration in immortalized human pancreatic duct epithelial cells and in human PDAC cell lines Panc-1 and BXPC-3 ( 34 ). In contrast, a Japanese laboratory published overexpression of GABA in 5 of 15 investigated PDAC tissue samples that were associated with overexpression of the pi subunit of the GABA A receptor ( 35 ). Differences in GABA contents in a typical Western diet as opposed to a typical Asian diet due to the high GABA contents in rice ( 36 ) as well as differences in smoking status may have contributed to the high GABA levels observed in PDACs by the Japanese group.
The significant reduction of GAD65 and GABA in xenografts of nicotine-treated animals in the current study suggests that nicotine suppresses GAD65 and that the associated reduction in GABA may contribute to the increased risk of smokers to develop PDAC. While the mechanisms responsible for these effects remain yet to be elucidated, a significant reduction in GABA has also been detected in the brain of smokers by proton magnetic resonance spectroscopy ( 16 ). It hence appears that nicotine may suppress the GABA system in multiple organs. This interpretation is consistent with the ubiquitous expression of nAChRs in mammalian cells ( 37 ) and the central regulatory role of this receptor family for the synthesis and release of the stress neurotransmitters and GABA ( 38 , 39 ). Interestingly, it has recently been shown that the tobacco carcinogen NNK, an agonist for nAChRs ( 40 , 41 ), reduced GABA levels in NNK-induced lung adenocarcinomas ( 42 ). These findings suggest that both nicotine and NNK may contribute to the development of PDAC in smokers by suppressing GAD and GABA.
Since the initial discoveries that nicotine stimulates the proliferation of small cell lung cancer cells ( 17 ) while inhibiting apoptosis ( 43 ), direct intracellular signaling via nAChRs in cancer cells has attracted considerable attention ( 22 , 44 ). However, a potential modulation of cancer development and progression via changes in systemic neurotransmitter levels that are regulated by nAChRs as observed in the current experiment has been largely overlooked. Disturbances in brain neurotransmission due to altered nAChRs in smokers have been associated with nicotine craving and addiction ( 45 ). In addition, many of the adverse cardiovascular effects of smoking are caused by nAChR-induced changes in the synthesis and release of adrenaline and noradrenaline ( 46 ). Our current findings suggest that this function of nAChRs may also have profound effects on the development and progression of numerous smoking-associated cancers. This interpretation is supported by a recent report that described a significant nicotine-induced growth stimulation of colon cancer xenografts associated with a systemic increase in adrenaline ( 47 ).
In summary, the current data suggest that the development and progression of PDAC is subject to significant modulation by changes in the levels of stimulatory stress neurotransmitters and inhibitory GABA and that GABA may be suitable for the prevention of PDAC in individuals at risk. With respect to the numerous factors in addition to smoking that can change the levels of these neurotransmitters and alter the sensitivity of their receptors, significant interindividual variations are to be expected. Attempts to utilize GABA for the prevention and treatment of PDAC should therefore be guided by the monitoring of biological markers such as systemic catecholamine and cAMP levels as well as tissue levels of GABA.
Funding
National Cancer Institute (RO1CA42829); State of Tennessee Center of Excellence for Human and Livestock Diseases.
Abbreviations
- ANOVA
analysis of variance
- cAMP
cyclic adenosine 3′,5′-monophosphate
- CREB
cyclic AMP response element-binding protein
- ERK
extracellular signal-regulated kinase
- GABA
γ-aminobutyric acid
- nAChR
nicotinic acetylcholine receptor
- NNK
4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone
- PDAC
pancreatic ductal adenocarcinoma
- PKA
protein kinase A
Conflict of Interest Statement: None declared.

{kind=link}
{kind=link}
{kind=link}
{kind=link}