




Abstract
The cytokine tumor necrosis factor (TNF) has pleiotropic functions both in normal physiology and disease. TNF signals by the virtue of two cell surface receptors, TNF receptor 1 (TNFR1) and TNF receptor 2 (TNFR2). Exogenous TNF promotes experimental metastasis in some models, yet the underlying mechanisms are poorly understood. To study the contribution of host TNFR1 and TNFR2 on tumor cell progression and metastasis, we employed a syngeneic B16F10 melanoma mouse model of lung metastasis combined with in vivo bioluminescence imaging. Treatment of tumor-bearing mice with recombinant human TNF resulted in a significant increase in tumor burden and metastatic foci. This correlated with an increase in pulmonary regulatory CD4 + /Foxp3 + T cells. TNF caused an expansion of regulatory T (Treg) cells in vitro in a TNFR2-dependent manner. To assess the contribution of immune cell expression of endogenous TNF and its two receptors on B16F10 metastasis, we generated bone marrow chimeras by reconstituting wild-type mice with bone marrow from different knockout mice. Loss of either TNF or TNFR2 on immune cells resulted in decreased B16F10 metastasis and lower numbers of Treg cells within the lungs of these animals. Selective depletion of Treg cells attenuated metastasis even in conjunction with TNF treatment. We propose a novel mechanism in which TNF activates TNFR2 on Treg cells and thereby expands this immunosuppressive immune cell population. Loss of either TNF or TNFR2 prevents the accumulation of Treg cells and results in a less tolerogenic environment, enabling the immune system to control B16F10 tumor metastasis and growth.
Introduction
Tumor metastasis is a complex multistep process requiring multiple interactions between tumor and host cells (parenchymal and endothelial cells as well as immune cells). Although some aspects of metastasis can be readily modeled and investigated in vitro , the genetic and biochemical determinants enabling a tumor cell to eventually form metastases mostly remain unclear ( 1 ). To improve cancer patient prognosis, it is of uttermost importance to shed light upon different molecular targets and to develop novel therapeutic strategies that could help to attenuate tumor cell metastasis.
The pleiotropic cytokine tumor necrosis factor (TNF) is mainly produced by activated immune cells, especially macrophages and T cells, but is frequently also expressed by tumor cells (reviewed in refs 2 and 3 ). TNF is a trimeric transmembrane type II protein from which a soluble form is released by proteolytic processing. Membrane-bound TNF and soluble TNF bind to two cell surface receptors, TNF receptor 1 (TNFR1) and TNF receptor 2 (TNFR2). There is also a homolog of TNF, lymphotoxin-α (LTα), which occurs as a soluble trimeric ligand that similar to soluble TNF interacts with both TNF receptors but in addition also to the related receptor herpes virus entry mediator. Membrane-bound TNF can strongly activate both TNF receptors, whereas soluble TNF only activates TNFR1 properly ( 4 ). It is evident from the analysis of TNFR1 and TNFR2 knockout (KO) mice that many immunoregulatory processes are controlled by the two TNF receptors in an antagonistic, additive or even synergistic way but there is also evidence for autonomous functions of TNFR1 and TNFR2. How the latter are realized at the molecular and cellular level, however, is largely unknown (reviewed in refs 3 and 5 ).
TNF was initially identified as a protein being able to induce tumor cell necrosis, hence the name ( 6 ). Indeed, recombinant TNF in combination with melphalan is in clinical use for the treatment of soft tissue sarcoma by isolated limb perfusion and leads to pronounced tumor necrosis in this scenario ( 7 ). It turned out, however, that necrosis induction in such established tumors was rather the result of TNF effects on tumor vasculature than by directly inducing tumor cell death ( 8 ).
A number of recent studies demonstrated a pro-tumorigenic role of the TNF/TNFR system in different inflammation-driven carcinogenesis models (reviewed in refs 2 and 3 ). Furthermore, early observations suggest that TNF appears to be a decisive factor in tumor metastasis. Stimulation of tumor cells with recombinant TNF in vitro ( 9–11 ), transduction of tumor cells with TNF ( 12 , 13) and treatment of mice with recombinant TNF before tumor cell inoculation ( 14–16 ), all enhanced the metastatic potential of the tumor cells in vivo . Likewise, exogenous and tumor cell-derived endogenous TNF enhanced pancreatic tumor growth and metastasis in an orthotopic mouse model ( 17 ). Only the latter of the above-mentioned studies looked at the contribution of endogenous TNF produced by non-transformed cells on tumor cell metastasis. Furthermore, it is unknown which TNFR needs to be activated on certain cell types in order to influence metastasis.
Experimental tumor cell metastasis is mostly described by the endpoint of tumor mass in the target organ after a defined period of time. Labeling tumor cells with firefly luciferase makes it possible to dynamically evaluate the tumor burden of each living animal by subjecting them to non-invasive bioluminescence imaging (BLI) ( 18–20 ). Here, we analyzed the role of TNF, TNFR1 and TNFR2 produced by immune cells on B16F10 lung metastasis and our results suggest that the TNF/TNFR system is involved in the homeostasis of regulatory T (Treg) cells and thereby affects the immunologic control of B16F10 metastases. In particular, immune cell-derived TNF appears to act on TNFR2 on Treg cells, expanding this immune cell subpopulation and thereby enhancing metastasis.
Material and methods
Cell culture medium and supplements were from Invitrogen (Darmstadt, Germany), all plasticware was from Greiner BioOne (Frickenhausen, Germany) and all antibodies were obtained from Biolegend (Uithoorn, The Netherlands) unless noted otherwise.
Animals
B6.TNF KO, B6.LT KO, B6.TNFR1 KO, B6.TNFR2 KO and B6.TNFR1/R2 KO (all in C57Bl/6 H-2 b background) mice were initially obtained from Jackson Laboratories (Bar Harbor, ME). Albino C57Bl/6 (Tyr<c-2J>) mice were a kind gift from Bettina Holtmann (Center for Experimental Molecular Medicine at the University Hospital Würzburg, Germany) and CD45.1 transgenic C57Bl/6 mice were a kind gift from Jörg Wischhusen (Department of Obstetrics and Gynecology, University Hospital Würzburg). B6.Foxp3.Luci.DTR-4 mice, expressing firefly luciferase, enhanced green fluorescent protein (eGFP) and the human diphtheria toxin (DTx) receptor under the control of the Foxp3 promoter ( 21 ) were a kind gift from Günter Hämmerling (German Cancer Research Center, Heidelberg, Germany). TNF, LTα, TNFR KO and B6.Foxp3.Luci.DTR-4 mice were backcrossed to albino C57Bl/6 to generate albino B6a.TNF KO, B6a.LTα KO, B6a.TNFR1 KO, B6a.TNFR2 KO, B6a.TNFR1/R2 KO and B6a.Foxp3.Luci.DTR-4 mice, which are superior to black mice in terms of in vivo BLI sensitivity. Genotypes of KO mice were routinely checked for by standard PCR. Female mice were used for experiments between 8 and 12 weeks of age. All mice were bred within the specified pathogen-free animal facility of the Center for Experimental Molecular Medicine at the Würzburg University Hospital receiving rodent chow and autoclaved drinking water ad libitum . All animal experiments were approved by local authorities (Regierung von Unterfranken) and complied with German animal protection law.
Generation of bone marrow chimeras
Female wild-type (WT) albino C57Bl/6 mice were lethally irradiated using a Faxitron CP-160 X-ray irradiation system (Faxitron X-Ray, Lincolnshire, IL) with a dose of 9 Gy and reconstituted with syngeneic bone marrow. To generate mice that were deficient for TNF, LTα or the TNFRs in their immune cells only, bone marrow cells were isolated either from female WT congenic CD45.1 transgenic B6 mice or from individual female KO mice. Donor mice were killed and femur and tibia bones were flushed with phosphate-buffered saline (PBS) to isolate bone marrow cells. The cell suspension was filtered through a 70 µm cell strainer (BD, Heidelberg, Germany) and 5×10 6 bone marrow cells in 100 µl PBS were injected intravenously into the retro-orbital venous plexus of irradiated host mice. Mice were treated with antibiotic drinking water (Baytril; Bayer, Leverkusen, Germany) for 1 week to prevent infections following myeloablative irradiation. After 7–10 weeks following reconstitution, mice were bled via their lateral tail vein and peripheral blood mononuclear cells were assessed for the expression of CD45.1 and CD45.2 by flow cytometry. Using CD45.1/2, as a surrogate marker, we found all mice to be 92.8±2.1% chimeric for donor immune cells (data not shown).
Cell culture
B16F10-Luc murine melanoma cells stably expressing firefly luciferase were maintained in Dulbecco’s modified Eagle’s medium supplemented with 10% fetal bovine serum, 1% antibiotics (penicillin and streptomycin), l -glutamine and 0.1% β-mercaptoethanol. Cells were trypsinized and passaged twice weekly. These cells are syngeneic to C57Bl/6 mice.
In vivo metastasis model and in vivo BLI
B16F10-Luc cells were trypsinized, harvested and washed twice with PBS. 100 000 cells were injected in 100 µl PBS via the lateral tail vein of C57Bl/6 mice. For TNF treatment, mice were injected intraperitoneal (i. p.) with 5 µg human TNF (hTNF) or 5 µg murine TNF (mTNF) in 200 µl PBS every other day starting on the day of tumor cell inoculation. For in vivo BLI on days 2, 4, 7, 9, 11 and 14 after tumor cell inoculation, mice were anesthetized with i. p. injection of 80mg/kg body wt esketamine hydrochloride (Pfizer, Berlin, Germany) and 16mg/kg body wt xylazine (cp Pharma, Burgdorf, Germany). Together with anesthetics, mice were injected with 300mg/kg body wt d -luciferin (Biosynth, Staad, Switzerland). Ten minutes later, bioluminescence signals of the anesthetized mice were assessed using an IVIS Spectrum imaging system (Caliper Life Sciences, Mainz, Germany). Pictures were taken both from ventral and dorsal view in automatic mode with a maximum exposure time of 5min per picture. Pictures were evaluated using Living Image 4.0 software (Caliper Life Sciences). A rectangular region of interest was drawn over the thorax of a mouse and aligned with its upper end to the ears. This region of interest was copied to all other mice of the experiment and aligned to their ears to guarantee that the same area was analyzed for all animals. Extrapulmonary metastases were deemed to be present when bioluminescence intensity was above background outside of the thorax region ( Supplementary Figure 1 , available at Carcinogenesis Online). For experiments with B6.Foxp3.Luci.DTR-4 mice, animals were inoculated with B16F10 cells not expressing luciferase and injected with 15ng DTx/g body wt (Sigma, Schnelldorf, Germany) on days −2, −1, 2, 4, 7 and 11 in respect to the day of tumor cell inoculation in order to deplete Treg cells.
Ex vivo imaging
On day 15 after tumor cell inoculation, mice were injected with d -luciferin and were euthanized 10min later. Internal organs were removed and subjected to ex vivo BLI ( 19 ). Pulmonary surface metastases of B16F10 cells were counted and tissue samples were embedded in Tissue Tek OCT (Sakura Finetek, Staufen, Germany) for further histological analysis or stored in PBS on ice before further processing for flow cytometry.
Isolation of immune cells from lung tissue and spleens
Lungs were minced with a surgical blade and digested for 30min at 37°C with 2mg/ml collagenase D and 0.1mg/ml DNase I (both from Roche, Mannheim, Germany). Tissue pieces were mashed through a 70 µm cell strainer and spun down. The cell pellet was resuspended in erythrocyte lysis buffer (168mM NH 4 Cl, 10mM KHCO 3 , 0.1mM ethylenediaminetetraacetic acid) and incubated for 2min. Next, 10vol of PBS were added and the cells were spun down again. The resulting pellet was resuspended in PBS and cells were used for flow cytometry. Spleens were directly filtered through a 70 µm cell strainer into erythrocyte lysis buffer and washed once with PBS.
Isolation of T cells and in vitro incubation with TNF
T cells were enriched from splenocytes of WT B6, B6.TNFR2 KO or B6.Foxp3.Luci.DTR-4 mice using the Dynal Mouse T cell Negative Isolation Kit (Invitrogen) according to the manufacturer’s instructions. 100 000 T cells per well were incubated with hTNF (0–100ng/ml) or mTNF (0–100ng/ml) in a 96-well round-bottom plate in RPMI medium supplemented with 10% fetal bovine serum, 1% antibiotics (penicillin and streptomycin), l -glutamine and 0.1% β-mercaptoethanol for 96h before being subjected to flow cytometry.
Flow cytometry
Cells were blocked with normal rat serum (1:20 in PBS) and stained with appropriate antibodies at 4°C for 30min. Following surface antigen staining, cells were washed once with PBS and labeled with propidium iodide. For intracellular staining, cells were stained with LIVE/DEAD Fixable Violet Dead Cell Stain Kit (Invitrogen) and further processed using the Mouse Regulatory T Cell Staining Kit #2 (eBioscience, Frankfurt, Germany) according to the manufacturer’s protocol. Following antibodies were used: CD25-PE (PC61.5) (eBioscience), CD4-FITC (RM4-5) (eBioscience), CD4-PE (RM4-5), CD45.1-PE (A20), CD45.2-APC (104), CD8-PE/Cy7 (53–6.7), Foxp3-APC (FJK-16s) (eBioscience) and NK1.1-FITC (PK136). All experiments were performed on a BD FACS Canto II (BD) and sample data were recorded using BD FACSDiva software and analyzed using FlowJo software (Tree Star, Ashland, OR).
Immunofluorescence microscopy
Cryo-embedded tissues were cut into 3 µm thick sections on a Leica CM1900 cryostat (Leica Microsystems, Wetzlar, Germany) and mounted onto frosted slides. Slides were air-dried and fixed with acetone at room temperature for 7min. Slides were washed and blocked with 2% fetal bovine serum in PBS for 15min. When biotin-conjugated antibodies were used, additional blocking using an Avidin/Biotin Blocking Kit (Vector Laboratories, Burlingame, CA) was performed. Slides were then incubated with the appropriate antibodies for 1h at room temperature. Between antibody incubations, the slides were washed with PBS thrice. In the end, slides were counterstained with 4′,6-diamidino-2-phenylindole and mounted with mounting medium (Vector Laboratories). Following antibodies were used: CD11b-Alexa 647 (M1/70), CD4-Alexa 647 (GK1.5), CD8-Biotin (53–6.7), Foxp3-purified (FJK-16s) (eBioscience), F4/80-Alexa 488 (A3-1), NKp46-Alexa 488 (29A1.4) (R&D Systems, Wiesbaden-Nordenstadt, Germany), donkey-anti-rat-Cy3 (Dianova, Hamburg, Germany), streptavidin-Alexa 546 (Invitrogen). Hematoxylin and eosin stainings were performed according to standard procedures. Images were obtained with a Zeiss Imager.Z1m fluorescence microscope (Carl Zeiss, Göttingen, Germany) and evaluated using Zeiss AxioVision software (Carl Zeiss). Immune cells within normal lung tissue and within B16F10 metastases were counted and given as cells per mm 2 . Metastases could be readily identified in the 4′,6-diamidino-2-phenylindole channel with tumor cells showing dysplastic nuclei and more light absorption due to high melanin production.
Statistics
All graphs shown are combined data from at least two independent experiments, except for Supplementary Figure 3 , available at Carcinogenesis Online; the number of animals is indicated in the figure legends. All data are shown as mean ± standard error of mean. Figures were prepared using GraphPad Prism 5 software (La Jolla, CA) and Adobe Photoshop 7 (San Jose, CA). All groups were compared with the WT or untreated control group, respectively, by non-parametric Mann–Whitney test using GraphPad InStat 3 software. Data reaching statistical significance are indicated as * P ≤ 0.05, ** P ≤ 0.01.
Results
Exogenous TNF enhances metastasis and induces Treg cells in the B16F10 tumor model
First, we wanted to reproduce earlier studies reporting that administration of exogenous TNF results in enhanced tumor cell metastasis ( 14–17 ). Using a luciferase-expressing variant of the B16F10 melanoma cell line and non-invasive in vivo BLI, we treated mice with 5 µg of hTNF every other day after tumor inoculation. In hTNF-treated mice, the signal emitted from the tumor cells was observed as early as day 2, whereas the untreated mice showed much less signal intensity at a later time point (day 7) ( Figure 1A ). Quantification of in vivo BLI pictures showed a 10-fold increase in light emission on day 14 after tumor cell inoculation in hTNF-treated mice ( Figure 1B ). Similarly, ex vivo imaging of the lungs derived from mice on day 15 showed an increase in signal intensity in the hTNF-treated group ( Figure 1C ). In the hTNF-treated group, the numbers of B16F10-Luc surface metastases significantly exceeded those observed in untreated mice ( Figure 1D ). Lung-infiltrating NK1.1 + , CD4 + and CD8 + cells did not differ between groups. However, Foxp3 + CD4 + Treg cells were 1.7-fold increased in the TNF-treated group ( Figure 1E ). Next, we compared the absolute numbers of CD8 + , CD4 + , Treg, CD11b + /F4/80 + and NKp46 + cells both within the B16F10-Luc tumors and non-tumor parenchymal lung tissue within the same untreated or hTNF-treated mice by fluorescence microscopy. Less CD8 + cells infiltrated tumor nodules of hTNF-treated mice in comparison with untreated mice ( Figure 1F ), whereas hTNF treatment increased the infiltration of CD11b + /F4/80 + macrophages into tumor nodules ( Supplementary Table 1 , available at Carcinogenesis Online). TNF treatment resulted in an increase of Treg cells within healthy lung tissue but not directly within tumor nodules ( Figure 1H ). hTNF treatment of B16F10-Luc tumor-bearing mice furthermore reduced the number of natural killer cells within normal lung tissue ( Supplementary Table 1 , available at Carcinogenesis Online). Taking into account that soluble hTNF only activates mouse TNFR1, but not TNFR2, we performed quantitative real -time-PCR for lung tissues from hTNF-treated and untreated mice. hTNF treatment significantly induced the expression of mTNF ( Supplementary Figure 2 , available at Carcinogenesis Online). We, furthermore, treated B16F10 tumor-bearing mice with mTNF and found the same effects as with hTNF treatment, that is, enhanced tumor metastasis and elevated numbers of pulmonary Treg cells ( Supplementary Figure 3 , available at Carcinogenesis Online).
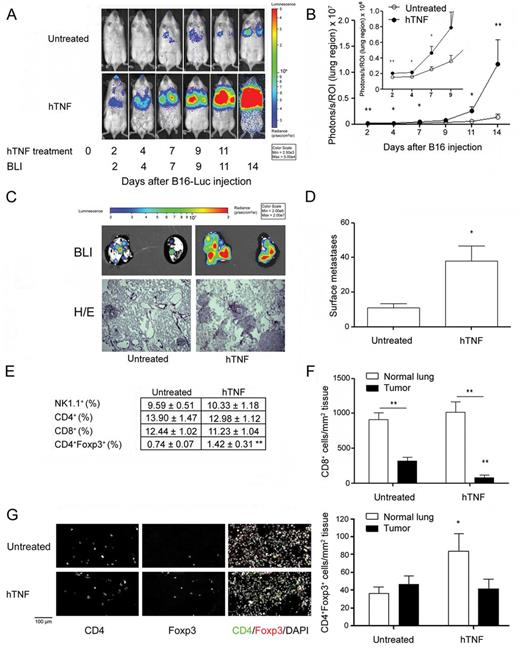
Exogenous hTNF strongly enhances B16F10-Luc metastasis. Female WT albino B6 mice were challenged with 1×10 5 B16F10-Luc tumor cells intravenosly and then either injected with 5 µg hTNF i. p. on days 0, 2, 4, 7, 9 and 11 or left untreated following the initial injection of tumor cells. ( A ) In vivo BLI of a representative mouse of each group. ( B ) Quantification of in vivo BLI images (photons/s of lung region); untreated n = 10, hTNF n = 8. ( C ) Upper row: Ex vivo BLI of a representative mouse of each group on day 15 after tumor cell inoculation; untreated n = 10, hTNF n = 8. Lower row: Representative histological sections of lungs showing B16F10-Luc metastases. ( D ) Numbers of pulmonary surface metastases (left lungs only); untreated n = 10, hTNF n = 8. ( E ) Flow cytometric analysis of immune cell subsets within lungs of TNF-treated or untreated mice; untreated n = 10, hTNF n = 6. ( F and G ) Immunofluorescence microscopy for the content of CD8 + cells (F), or Treg cells (G) within tumor nodules and normal lung tissue of B16F10-Luc tumor-bearing female untreated and hTNF-treated mice. One exemplary photomicrograph is shown for normal lung tissue for both groups. Treated mice were compared withuntreated mice for both normal lung tissue and tumors, and for each group, normal lung tissue was compared with tumors (animals/metastases; animals/fields of normal tissue): untreated n = 4/8; 5/6; hTNF n = 3/9; 3/5. Mean ± standard error of the mean, * P ≤ 0.05, ** P ≤ 0.01.
mTNF, but not hTNF, induces Treg cells in vitro in a TNFR2-dependent manner
We next tested the effect of TNF on Treg cells in vitro and by which TNFR an anticipated expansion of this immune cell subset is mediated. Isolated splenic T cells from B6.Foxp3.Luci.DTR-4 mice were treated with recombinant soluble hTNF, which only binds to mTNFR1, and recombinant mTNF, which binds to both mTNF receptors. As TNFR2 in contrast with TNFR1 is only poorly responsive to soluble TNF, we used high concentrations of TNF for stimulation. mTNF, but not hTNF, induced Treg cells after 96h of incubation ( Figure 2A ). Loss of TNFR2 on T cells completely abrogated the inductive effect of high concentrations of mTNF on Treg cell numbers ( Figure 2B ). Furthermore, we found TNFR2-deficient mice to have only half as much Treg cells among splenic T cells as WT mice.
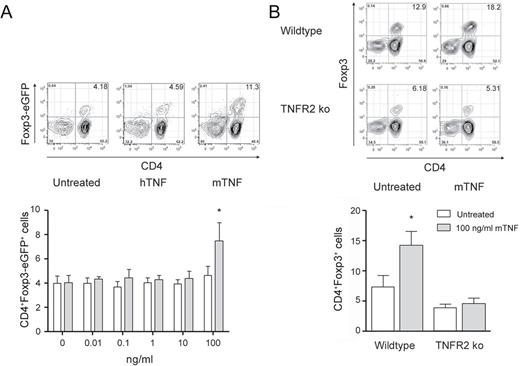
mTNF, but not hTNF, induces Treg cells in vitro in an TNFR2-dependent manner. ( A ) 100 000 B6.Foxp3.Luci.DTR-4 T cells were cultured with hTNF or mTNF for 96h in vitro as indicated or left untreated before flow cytometric analysis for CD4 + Foxp3-eGFP + expressing cells. Shown are representative cytometric data for each group (untreated, 100ng/ml hTNF, 100ng/ml mTNF) and the respective data analysis ( n = 5). ( B ) 100 000 B6 WT or B6.TNFR2 KO T cells were cultured with 100ng/ml mTNF for 96h in vitro or left untreated before flow cytometric analysis for CD4 + Foxp3 + expressing cells. Shown are representative cytometric data for each group and the respective data analysis ( n = 5). Mean ± standard error of the mean, * P ≤ 0.05.
Immune cell-restricted deficiency of TNF or TNFR2 leads to decreased B16F10-Luc metastasis and reduced numbers of Treg cells
Mice deficient of components of the TNF receptor family pathways display perturbations in lymphoid organ and immune cell development and might show elevated levels of circulating TNF in the absence of TNFRs ( 22–24 ). These changes in constitutive KO mice may, therefore, strongly bias B16F10-Luc tumor growth, rather than being a direct consequence of TNF/LTα–TNFR interactions. To circumvent this potential bias and to elucidate the effects of TNF, LTα and TNF receptor deficiency in immune cells only, we generated bone marrow chimeras.
Neither LTα KO nor TNFR1 KO bone marrow chimeras displayed differences in tumor signals compared with WT bone marrow chimeras. However, TNF KO or TNFR2 KO chimeras displayed significantly less in vivo BLI signals compared with the control group ( Figure 3A and B ; data not shown). This effect was also seen by ex vivo imaging ( Figure 3C ), and histological analysis, furthermore, showed less metastases in these two groups ( Figure 3D ). B16F10-Luc surface metastases were significantly reduced in the TNFR2 KO bone marrow chimeras compared with the WT chimeras ( Figure 3E ). Furthermore, TNF KO and TNFR2 KO chimeras displayed fewer extrapulmonary metastases than WT chimeras (WT → WT 4/13 [30.8%], TNF KO → WT 1/11 [9.1%], LTα KO → WT 3/11 [27.3%], TNFR1 KO → WT 4/10 [40%], TNFR2 KO → WT 1/10 [10%]). More Treg cells (2.02±0.48%) resided in the lungs of TNFR1 KO chimeras in contrast with fewer Treg cells (0.80±0.27%) in TNFR2 KO chimeras when compared with WT chimeras (1.37±0.30% Treg cells), although these differences deemed not statistically significant ( Figure 3F ).
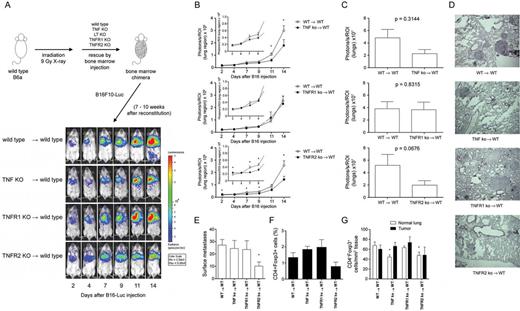
Loss of TNF or TNFR2 in immune cells decreases B16F10-Luc metastasis and reduces Treg cells. In vivo and ex vivo BLI of female bone marrow chimeric mice ( A ) Female WT mice were lethally irradiated and reconstituted with 5×10 6 bone marrow cells isolated from female CD45.1 + WT mice, TNF KO, TNFR1 KO and TNFR2 KO mice. After 7–10 weeks following reconstitution, mice were injected with 1×10 5 B16F10-Luc tumor cells and imaged at the days after tumor cell inoculation as indicated. ( B ) Quantification of in vivo BLI images (photons/s of lung region). KO-reconstituted mice were compared with the WT-reconstituted group; WT → WT n = 10, TNF KO → WT n = 11, TNFR1 KO → WT n = 10, TNFR2 KO → WT n = 10. ( C ) On day 15 after tumor cell inoculation, animals were killed and light emission from lungs was quantified by ex vivo BLI. KO-reconstituted mice were compared with the WT-reconstituted group; WT → WT n = 10, TNF KO → WT n = 11, TNFR1 KO → WT n = 10, TNFR2 KO → WT n = 10. ( D ) Representative histological sections of lungs showing B16F10-Luc metastases. ( E ) Number of pulmonary surface metastases (left lungs only); WT → WT n = 6, TNF KO → WT n = 5, TNFR1 KO → WT n = 6, TNFR2 KO → WT n = 5. ( F ) Fluorescence-activated cell sorting analysis for the content of Treg cells within lungs of tumor-bearing female WT-reconstituted and KO-reconstituted mice. WT → WT n = 6, TNF KO → WT n = 5, TNFR1 KO → WT n = 6, TNFR2 KO → WT n = 5. ( G ) Immunofluorescence microscopy for the content of immune cells within tumor nodules and normal lung tissue of B16F10-Luc tumor-bearing female bone marrow chimeric mice. KO-reconstituted mice were compared with WT-reconstituted mice (animals/metastases; animals/fields of normal tissue): WT → WT n = 9/19, 9/12; TNF KO → WT n = 8/17, 6/8; TNFR1 KO → WT n = 3/9, 6/9; TNFR2 KO → WT n = 6/9, 4/7. Mean ± standard error of the mean, * P ≤ 0.05.
In chimeras, fewer CD8 + and NKp46 + cells infiltrated into B16F10-Luc tumors than into normal lung tissues from the same animals; for macrophages, this was reversed and these cells accumulated within B16F10 metastases within all genotypes ( Supplementary Figure 4 , available at Carcinogenesis Online). In both TNF KO and TNFR2 KO chimeras, we found fewer pulmonary Treg cells than in WT chimeras ( Figure 3G ).
Depletion of Treg cells attenuates B16F10 metastasis irrespective of exogenous TNF treatment
We found hTNF-induced B16F10 metastasis to be associated with an increase in Treg cells and therefore wanted to further elucidate the involvement of this T-cell subset. To this end, we used transgenic B6.Foxp3.Luci.DTR-4 mice that express firefly luciferase, eGFP and the DTx receptor under the Foxp3 promoter in Treg cells ( 21 ). To deplete Treg cells, we treated these animals with DTx before the inoculation with B16F10-Luc cells and treatment with hTNF. In vivo and ex vivo imaging revealed a significant reduction in luciferase signals indicating depletion of luciferase + Treg cells (data not shown; Figure 4A and B ). Flow cytometry of the lungs confirmed significantly reduced numbers of eGFP-expressing Treg cells ( Figure 4C ). Both effector CD4 + T cells and Treg cells expressed little TNFR1. Approximately, 30% of pulmonary Treg cells expressed TNFR2 in contrast with only 10% TNFR2 + effector CD4 + T cells ( Figure 4D ). DTx-treated animals exhibited significantly reduced numbers of B16F10 surface metastases ( Figure 4E ) in comparison with untreated control mice. The numbers of pulmonary metastases increased in B6.Foxp3.Luci.DTR-4 mice treated with hTNF ( Figure 4E ) and correlated with significantly increased lung-infiltrating Treg cells ( Figure 4C ). TNF treatment did not change the expression pattern of TNFR1 and TNFR2 on CD4 + T cells ( Figure 4D ). Treatment of B6.Foxp3.Luci.DTR-4 mice with both DTx and hTNF reversed the inductive effect of hTNF on B16F10 metastasis and numbers of pulmonary metastases did not differ from untreated control mice ( Figure 4E ). To rule out a direct effect of DTx on B16F10 cells, we also treated WT mice with DTx. Treatment with DTx alone neither showed significant effect on metastasis numbers nor was DTx directly cytotoxic to B16F10 cells in vitro (data not shown). We were interested to know whether human tumor-infiltrating Treg cells express TNFR2 and thus stained colon adenocarcinoma specimens for Foxp3 and TNFR2. Most Treg cells expressed TNFR2 in these patient samples ( Supplementary Figure 5 , available at Carcinogenesis Online).
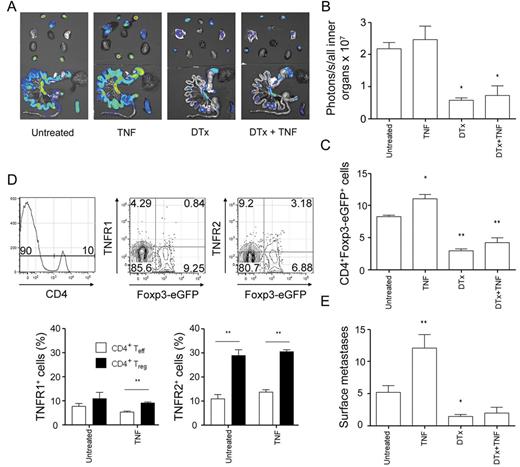
Depletion of Treg cells attenuates B16F10 metastasis irrespective of hTNF treatment. ( A ) B6.Foxp3.Luci.DTR-4 mice were injected with two i. p. doses of 15ng DTx/g body wt on days −2, −1 with respect to intravenous tumor cell inoculation in order to deplete Treg cells. Treg depletion was held up with consecutive DTx injections on days 2, 4, 7 and 11. Mice were injected with 5 µg hTNF i. p. on days 0, 2, 4, 7, 9 and 11 or left untreated. Ex vivo BLI of a representative B6.Foxp3.Luci.DTR-4 mouse of each group. ( B ) Quantification of ex vivo BLI; untreated n = 8, hTNF n = 6, DTx n = 4, DTx + hTNF n = 4. ( C ) Fluorescence-activated cell sorting analysis for eGFP-expressing cells within CD4 + compartment; untreated n = 6, hTNF n = 6, DTx n = 10, DTx + hTNF n = 6. ( D ) Fluorescence-activated cell sorting analysis for the expression of TNFR1 and TNFR2 on CD4 effector and Treg cells; untreated n = 8, hTNF n = 6. ( E ) Number of pulmonary surface metastases (left lungs only); untreated n = 17, hTNF n = 9, DTx n = 13, DTx + hTNF n = 4. Mean ± standard error of the mean, * P ≤ 0.05, ** P ≤ 0.01.
Discussion
TNF was initially described to exhibit anticarcinogenic functions ( 6 ) with newer studies demonstrating promotional effects in inflammation-induced carcinogenesis (reviewed in refs 2 and 3 ) and tumor metastasis ( 14–17 ). A number of target cells and mechanisms were proposed to be involved in the pro-metastatic effects of TNF including cellular adhesion, invasion and inflammation ( 9–11 , 17 , 25 , 26 ).
In this study, we used an in vivo BLI-based mouse model to assess the contributions of TNF, LTα and their cell surface receptors TNFR1 and TNFR2 to B16F10 murine melanoma cell metastasis with a special focus on the involvement of immune cells. In concordance with earlier studies, treatment of mice with recombinant hTNF or mTNF massively enhanced B16F10-Luc metastasis, which was accompanied with reduced infiltration of CD8 + T cells into tumor nodules within the lungs of treated animals. It was proposed by Sauer et al. (27) that CD8 + T cells play an important role in B16F10 tumor cell metastasis and loss of these immune cells enhances the formation of pulmonary tumor nodules. The authors found recombinant mTNF to be directly cytotoxic to B16F10 cells in vitro and proposed CD8 + T-cell-derived TNF to act antimetastatic in this model. Though there was an association between decreased infiltration of CD8 + T cells into tumor nodules and the extent of tumor metastasis in this study, we could not find recombinant mTNF to be cytotoxic to B16F10 cells in comparable concentrations as in the aforementioned study (data not shown).
Very little is known about the contribution of endogenous TNF or the two TNFRs to tumor cell metastasis. To address this, we performed B16F10-Luc metastasis experiments using mice deficient for TNF, LTα, TNFR1 or TNFR2 within the hematopoietic compartment. Neither the loss of LTα nor TNFR1 modulated B16F10-Luc metastasis, whereas the loss of either TNF or TNFR2 significantly reduced metastasis.
Orosz et al. ( 14 ) treated mice with either recombinant mTNF or hTNF before the intravenous inoculation of CFS1 cells and found an increase in lung metastases for both TNF variants. However, the effect was more pronounced for mTNF than for hTNF. Due to the inability of hTNF to bind to mTNFR2, these results indicate that activation of TNFR2 is more crucial for TNF-induced metastasis than activation of TNFR1 in this model ( 28 ). As TNF itself is a target gene of TNFR1-induced signaling pathways, hTNF has the potential to induce mTNF and can thus trigger TNFR2 activation indirectly (( 29 ) and our own data). On the other hand, employing the colon 26 metastasis model, it has been shown that TNFR1 can have a crucial role in hepatic metastasis ( 30 ). Sasi et al. ( 31 ) assessed the growth of subcutaneously implanted Lewis lung carcinoma and B16F10 cells in WT and TNFR2 KO mice and found decreased tumor sizes within the latter group. The authors concluded from this study that TNFR2 is involved in tumor angiogenesis and that the loss of this receptor results in less vascular endothelial growth factor expression and reduced capillary densities within the tumor tissue. The tumors behaved the same in TNFR1/R2 double-KO mice and in WT mice. Although the study by Sasi et al. (31) aimed at the effects of the TNF–TNFR system on the growth of solid tumors only, rather than on metastasis, the results might point toward a novel mechanism of how TNF promotes tumor growth ( 31 ). Charles et al. ( 32 ) determined the effects of TNF signaling in a mouse model of ovarian cancer. The loss of TNFR1, but not of TNFR2, on immune cells resulted in reduced tumor growth. TNFR1-dependent production of interleukin-17 by CD4 + cells and subsequent recruitment of myeloid cells into the tumor was discussed to be the underlying mechanism of how TNF enhances tumor growth in this model. It seems clear from the above-mentioned studies that TNF modulates tumor growth in a number of different manners depending on the tumor itself and its microenvironment.
Treatment of B16F10 tumor-bearing mice with exogenous TNF in our experiments resulted in increased numbers of pulmonary Treg cells, whereas the loss of TNFR2 decreased Treg cell numbers. These effects correlated with tumor cell signals in both settings. TNF induces Treg cells in vitro in a TNFR2-dependent manner and the depletion of Treg cells significantly reduced the number of pulmonary surface metastases and also ablated the enhancing effect on B16F10-Luc metastasis that was seen with TNF treatment underlining the importance of this T-cell subset in this experimental setting.
Treg cells have been discussed to play an important role in the growth and metastasis of some cancer variants and it could be shown that experimental depletion of these cells reduces the expansion and spread of tumor cells ( 33–37 ). We found that about 30% of pulmonary Treg cells express TNFR2, whereas only about 10% of effector CD4 + T cells do so. Furthermore, TNFR1 expression on Treg cells was low in comparison with TNFR2 expression. A number of studies describe the preferential expression of TNFR2 on murine and human CD4 + CD25 + Foxp3 + Treg cells and claim this to be a decisive factor for the suppressive activity of these cells. Nevertheless, the functional outcome of this observation is conflicting to date. Some studies found this TNFR2 + Treg subset to exhibit higher suppressive activity than TNFR2 − Treg cells ( 38–41 ), whereas other studies proposed the opposite, that is, TNF reduces Treg functions by activating TNFR2 ( 42–44 ). TNF is generally regarded to mainly exhibit pro-inflammatory functions. The observation that TNFR2 expression on Treg cells correlates with their suppressive activity indicates a self-induced negative feedback loop for the actions of TNF. Chen et al. ( 38 ) demonstrated in the Lewis lung carcinoma model that the majority of tumor-infiltrating Treg cells are positive for TNFR2 expression and that these cells are more suppressive than Treg cells isolated from lymph nodes of tumor-bearing mice. TNFR2 is also upregulated on tumor-infiltrating conventional CD4 T cells. This was demonstrated to enable conventional T cells to resist suppression by TNFR2-negative, but not TNFR2-positive Treg cells, strengthening the idea that TNFR2 on Treg cells is a decisive factor in terms of their suppressive functions ( 45 ). The observation that TNFR2 + Treg cells accumulate within tumors might set the biological basis for an expansion of these cells by TNF treatment as seen in our own experiments and also help explain why TNFR2 KO mice, which have reduced basal numbers of Treg cells, exhibit less B16F10 tumor growth. We not only found mouse Treg cells to express TNFR2 but also tumor-infiltrating Treg cells in human colon adenocarcinoma specimens.
The data presented in this study suggest that TNF activates TNFR2 on Treg cells and thereby expands this immune cell subset. This generates a more tolerogenic environment favoring the outgrowth of B16F10-Luc metastases. Loss of either TNF or TNFR2 on immune cells results in the attenuation of B16F10 metastasis accompanied by lower pulmonary Treg numbers. Depletion of Treg markedly decreases metastasis and also impedes the pro-metastatic effect of TNF. This is the first time that the control of Treg homeostasis by TNF could be linked to the pro-metastatic effect of this pleiotropic cytokine.
Supplementary material
Supplementary Table 1 and Figures 1–5 can be found at Supplementary Data
Funding
Interdisziplinäre Zentrum für Klinische Forschung der Universität Würzburg (grant B-149); and the Deutsche Forschungsgemeinschaft (grant SFB-TR 52 Z2 and DFG Wa 1025/18-1).
Conflict of Interest Statement: None declared.
Acknowledgement
We acknowledge the expert technical assistance of Claudia Pregitzer (Department of Pathology, Würzburg University, Würzburg, Germany) and Jeanette Baker and all members of the Beilhack laboratory for discussion and critical review of this manuscript.
Abbreviations:
- BLI
bioluminescence imaging
- DTx
diphtheria toxin
- eGFP
enhanced green fluorescent protein
- hTNF
human TNF
- i. p.
intraperitoneal
- KO
knockout
- LTα
lymphotoxin-α
- mTNF
murine TNF
- PBS
phosphate-buffered saline
- TNF
tumor necrosis factor
- TNFR1
TNF receptor 1
- TNFR2
TNF receptor 2
- Treg
regulatory T
- WT
wild-type.
References

{kind=link}
{kind=link}
{kind=link}
{kind=link}