




Abstract
Dietary inadequacy of folate enhances and folate supplementation suppresses colorectal carcinogenesis in the dimethylhydrazine rat model. Folate is an essential factor for DNA methylation and the de novo biosynthesis of nucleotides, aberrations of which play important roles in mutagenesis. This study investigated whether the mutational hot spots of the Apc and p53 genes for human colorectal cancer are mutated in dimethylhydrazine-induced colorectal neoplasms and whether dietary folate can modulate mutations in these regions. Rats were fed diets containing 0, 2 (basal requirement), 8 or 40 mg folate/kg diet. Five weeks after diet initiation, dimethylhydrazine was injected weekly for 15 weeks. Mutations were determined by direct sequencing in 11 low and seven high grade dysplasias and 13 invasive adenocarcinomas. A total of six Apc mutations were found in four dysplastic and carcinomatous lesions: two in two low grade dysplasias, two in one high grade dysplasia and two in one adenocarcinoma. All mutations were single base substitutions, four of which were A:T→G:C transitions. Five of the six mutations were located upstream from the region corresponding to the human APC mutation cluster region. Dietary folate had no effect on the frequency and type of Apc mutations. No mutations were detected in exons 5–9 of the p53 gene in neoplastic lesions. These data suggest that in the dimethylhydrazine rat model of colorectal cancer, the Apc gene is mutated in early stages, albeit to a lesser degree than observed in human colorectal cancer, whereas the mutational hot spot of the p53 gene for human colorectal cancer is not commonly mutated. Although the low frequency of Apc mutations and the small number of neoplasms studied in this study might have precluded our ability to observe modulatory effects of folate, dietary folate appears to have no significant effect on Apc and p53 mutations.
Diminished folate status, assessed by either dietary intake or by the measurement of blood folate concentrations, has been associated with an increased risk of colorectal neoplasia (reviewed in refs 1,2). Collectively, epidemiological studies suggest an ~40% reduction in the risk of colorectal neoplasms in individuals with the highest dietary folate intake compared with those with the lowest intake ( 1 , 2 ). Animal studies, performed in the dimethylhydrazine (DMH) rat model of colorectal cancer (CRC), have also provided considerable support for a cause–effect relationship between folate deficiency and colorectal carcinogenesis as well as a dose-dependent protective effect of modest levels of dietary folate supplementation above the basal dietary requirement ( 3 , 4 ).
To date, the mechanisms by which folate can modulate colorectal carcinogenesis have not been clearly elucidated. Among the proposed mechanisms ( 1 , 2 ) is a pro-mutagenic effect of folate deficiency due to a disruption in the transfer of one-carbon moieties, the sole biochemical function known for folate ( 5 ). Folate is an essential factor for the de novo biosynthesis of purines and the pyrimidine thymidylate ( 5 ). The fidelity of DNA replication and repair is dependent on the correct balance and availability of the deoxynucleotide precursors (dNTPs) ( 6 ). Folate deprivation causes an imbalance in the ratios of the dNTPs for DNA synthesis ( 7 – 9 ). A chronic imbalance in dNTP ratios has been shown to be pro-mutagenic in vivo and in vitro and enhances sensitivity to DNA-damaging effects of carcinogens ( 10 ). Folate is also involved in the synthesis of methionine, which is a precursor of S -adenosylmethionine, the methyl donor for most biological methylation reactions, including that of DNA ( 5 ). DNA methylation at cytosine residues in cytosine–guanine doublets (CpG) is considered to play an important role in mutagenesis ( 11 , 12 ). CpG sequences within certain genes (e.g. the APC and p53 tumor suppressor genes) are mutational hot spots for several cancers, including CRC ( 13 – 15 ). The majority of mutations observed in CpG sequences within these genes are C→T transitions ( 13 – 15 ). Both methylated and unmethylated cytosines in CpG dinucleotides have been observed to be hypermutable via several different mechanisms ( 16 , 17 ). DNA hypomethylation has also been observed to increase mutation rates at non-CpG sites via genomic instability ( 18 ). Previously, dietary folate has been shown to modulate CpG methylation in the p53 gene in a site-specific manner ( 19 , 20 ).
Although the pro-mutagenic effect of folate deficiency, either alone or in response to alkylating agents, has been demonstrated in cell culture systems ( 8 , 21 , 22 ), this effect is yet to be determined in the intact animal. The present study investigated whether the mutational hot spots of the Apc and p53 genes for human CRC are also mutated in a DMH rat model of CRC in the setting where manipulation of dietary folate was used to modulate colonic carcinogenesis and whether dietary folate can modulate mutations in these regions. The Apc and p53 genes are two commonly mutated genes in early and late stage human colorectal carcinogenesis, respectively ( 23 ). About 60% of the somatic mutations of the APC gene in human CRC are clustered in a 700 bp region in exon 15, designated the mutation cluster region (MCR) ( 13 , 14 ). The majority of p53 mutations occur within a highly conserved area spanning from codon 110 to 307 (exons 5–8) ( 15 ). Up to 50% of somatic APC mutations within the MCR and of p53 mutations in exons 5–8 in human CRC are C→T transitions occurring at CpG sites ( 13 – 15 ).
The present study was part of a larger experiment designed to determine the protective effect of different levels of dietary folate on the development of colorectal neoplasms in DMH-treated rats. The detailed protocol has been published previously ( 4 ). Briefly, 80 weanling male Sprague–Dawley rats (60–90 g; Charles River Co., Wilmington, MA) were randomly assigned to receive an amino acid defined diet (Dyets, Bethlehem, PA) ( 24 ) containing either 0, 2 (basal dietary requirement for the rat), 8 or 40 mg folate/kg diet ( n = 20 for each group). Diets and water were supplied ad libitum . Body weights were recorded weekly. Five weeks after diet initiation, weekly s.c. injections of 44 mg/kg body weight DMH base (Sigma Chemical Co., St Louis, MO) were begun and then continued for 15 weeks. The negative control rats ( n = 5) were fed the identical diet containing 2 mg folate/kg diet for the same duration as the DMH-treated rats but were not injected with DMH. The rats were killed 15 weeks after the first injection of DMH (i.e. after 20 weeks of the defined diets) by exsanguination under carbon dioxide anesthesia.
At the time of death, blood was withdrawn from the inferior vena cava using a pre-heparinized 18 gauge needle into vacutainer tubes containing EDTA and centrifuged at 800 g for 10 min at 4°C. Plasma was stored at –70°C in 0.5% ascorbic acid for the plasma folate assay.
The colorectum was excised and put on a glass plate suspended on crushed ice. The colorectum was opened longitudinally, rinsed in 0.9% NaCl and examined for any macroscopic lesions under a dissecting microscope. Macroscopic lesions were identified and recorded. The colorectum was then sectioned longitudinally into two halves. The mucosal layer from one half was carefully scraped using glass slides. The resulting mucosal scrapings were rapidly weighed, frozen in liquid nitrogen and stored at –70°C for subsequent analysis of colonic mucosal folate concentrations. The other half was rolled up in `Swiss roll' fashion, fixed in 10% buffered formalin and embedded in paraffin ( 25 ). A single longitudinal section (5 μm) was cut from the midline of each roll and stained with hematoxylin and eosin (H&E). The Swiss roll is a means by which an intact longitudinal section of the entire length of the colorectum can be presented on a single microscope slide ( 25 ). Since midline sections are cut without regard to the presence of macroscopic lesions, each 5 μm section constitutes a random and representative longitudinal section from the entire length of the colon ( 25 ). This method has been used previously to quantitatively and qualitatively describe DMH-induced lesions in the colorectum ( 3 , 4 , 25 ). Histological lesions in the colonic mucosa were classified into normal, low grade dysplasia (LGD), high grade dysplasia (HGD) and invasive adenocarcinoma (CA) according to previously described criteria ( 26 ) by three gastrointestinal pathologists in blind fashion.
Plasma and colonic mucosal folate concentrations were measured by a microtiter plate assay using Lactobacillus casei as previously described ( 27 ). Five micrometer thick sections were examined by H&E staining and areas corresponding to microscopic lesions were marked on the matched unstained specimens. DNA from each microscopic lesion was extracted as crude preparations using proteinase K lysis mix (10 mM Tris–HCl, pH 8.0, 100 mM KCl, 2.5 mM MgCl 2 , 0.45% Tween 20 and 1 mg/ml proteinase K) as previously described ( 28 ). Care was taken to avoid contamination from adjacent non-neoplastic tissues. The sections were homogenized in the lysis mix and digested for 1 h at 65°C followed by 10 min at 95°C. Extracted DNA was stored at –20°C until mutation analysis was performed. Colonic DNA from normal and negative controls was extracted from areas corresponding to normal histology from paraffin blocks in a similar fashion ( 28 ).
A 2704 bp region, between nt 2074 and 4778 in exon 15 of the Apc gene, including the MCR (nt 3906–4589), was amplified by PCR using three pairs of exon primers to generate three overlapping segments (segment A, nt 2074–3061; segment B, nt 2908–3970; segment C, nt 3853–4778). The primer sequences were constructed based on the published rat Apc cDNA sequence (29; GenBank accession no. D38629) and synthesized by the ACGT Corp. (Toronto, Canada). The sequences of the primers were as follows.
Segment A:sense,5′-ATAGTCAGTAATGCATGTGGAACTCTGTGG-3′;
antisense,5′-CTAGGTCGGCAGGATACTGCCCATAACTGC-3′.
Segment B:sense,5′-GTGGAATATAAGAGATCTTCAAATGACAGG-3′;
antisense,5′-CTTTTATCTCTGCTATTTGCAGAGTATTCG-3′.
Segment C:sense,5′-TGTTTCTCAAGGTGTAGTTCCTTATCATCG-3′;
antisense,5′-TCTTCTAATATTTCAATATCATCATCATCG-3′.
Each 5.0 μl of DNA sample was amplified by PCR in a 50 μl volume containing 100 pmol of each primer, 0.25 mM each dNTP, PCR buffer (Qiagen, Mississauga, Canada), 1.5 mM MgCl 2 and 2 U of HotStart Taq polymerase (Qiagen). Following hot start PCR at 95°C for 5 min, 35 cycles of denaturation (95°C) for 1 min, annealing (54°C) for 1 min and extension (72°C) for 1 min were performed in a thermal cycler (PTC-200 DNA Engine ® ; MJ Research, Watertown, MA). All PCR amplifications included a 10 min extension at 72°C after cycle 35.
Exons 5, 6–7 and 8–9 of the p53 gene were amplified by PCR using three pairs of intron primers. Intron primer sequences for each exon of the rat p53 gene have been published ( 30 ) and were as follows.
Exon 5:sense:5′-GACCTTTGATTCTTTCTCCTCTCC-3′;
antisense,5′-GGGAGACCCTGGACAACCAG-3′.
Exons 6–7:sense:5′-CTGGTTGTCCAGGGTCTCC-3′;
antisense,5′-CCCAACCTGGCACACAGCTT-3′.
Exons 8–9:sense:5′-CTTACTGCCTTGTGCTGTGC-3′;
antisense,5′-CTTAAGGGTGAAATATTCTCC-3′.
The use of intron primers obviates amplification of p53 pseudogenes present in the rat ( 31 ). All primers were synthesized by Life Technologies (Gaithersburg, MD). Each 2.5 μl of DNA sample was amplified by PCR in a 50 μl volume containing 50 pmol of each primer, 0.2 mM each dNTP, PCR buffer (Perkin Elmer Cetus, Foster City, CA), 2.0 mM MgCl 2 and 2.5 U of Ampli Taq DNA polymerase (Perkin Elmer Cetus). Following hot start PCR, 30 cycles of denaturation (94°C) for 1 min, annealing (52°C for exon 5, 60°C for exons 6–7 and 56°C for exons 8–9) for 1 min and extension (72°C) for 1 min were performed in a thermal cycler (model 9600; Perkin Elmer Cetus).
The PCR products for the Apc (segments A–C) and p53 (exons 5–9) genes were gel purified using the Qiaex II Agarose Gel Extraction Kit (Qiagen) according to the manufacturer's protocol, re-extracted and dissolved in 50 μl of ddH 2 O. Each PCR product was then reamplified using nested primers under the same PCR conditions as for the Apc and p53 genes, respectively, gel purified, re-extracted and dissolved in 50 μl of ddH 2 O.
Coding strands of segments A, B and C within exon 15 of the Apc gene and exon 5, exons 6–7 and exons 8–9 of the p53 gene from all purified PCR products were directly sequenced using the Dideoxy Terminator Label Cycle Sequencing Kit (Applied Biosystems, Foster City, CA) and an Applied Biosystems 373 Sequencer as previously described ( 32 ). For the Apc gene, nested primers for direct sequencing were constructed based on the published rat Apc cDNA sequence (29; GenBank accession no. D38629). For the p53 gene, the published sequences of nested intron and flanking primers ( 30 ) were used for direct sequencing. All primers were synthesized by either the ACGT Corp. or Life Technologies. The Apc and p53 sequences thus generated were analyzed against the GenBank sequences [ Apc , accession no. D38629 ( 29 ); p53 , accession no. L07781 ( 31 )] using the DNASIS software program (Hitachi, San Diego,CA). Mutations were confirmed by directly sequencing the opposite strand with an automated sequencer as above and by subcloning PCR products into pBluescript II KS(+) vector (Stratagene, Cambridge, UK) and directly sequencing the plasmids with inserts using a T7 DNA Sequencing Kit (Amersham Pharmacia Biotech, Quebec, Canada) and [α- 35 S]dATP (Amersham Pharmacia Biotech).
Differences among groups were determined by one-way analysis of variance. Fisher's least significant difference (LSD) test was used for multiple comparisons. Pearson's coefficient of regression model was used to assess a correlation between plasma and colonic folate concentrations and dietary folate levels. For categorical response variables, differences among the groups were assessed by Fisher's exact test. Statistical analyses were performed using SYSTAT 6 for the Macintosh (Systat, Evanston, IL). All significance tests were bilateral and the significance level was set at 0.05. Results are expressed as mean ± SEM.
Previously, the larger study in which the present study was conducted demonstrated that increasing dietary folate up to four times the basal requirement leads to a progressive reduction in the evolution of macroscopic neoplasms from microscopic foci in a dose-dependent manner and that folate supplementation beyond four times the requirement does not convey further benefit ( 4 ). In the present study, a total of 31 microscopic lesions (11 LGD, seven HGD and 13 CA) from 31 rats with diagnostic agreement by the three pathologists were processed for mutation analysis. Colonic DNA from 10 rats whose colon was free of any microscopic lesions were chosen as normal. The negative control rats ( n = 5) were fed the identical diet containing 2 mg folate/kg diet for the same duration as the DMH-treated rats but were not injected with DMH. The proportions of normal, LGD, HGD and CA chosen for the present study were not significantly different among the four dietary groups (Table I ).
Growth curves were similar in the five groups of rats. The mean plasma and colonic mucosal folate concentrations were different among the four DMH-treated groups ( P < 0.001) and correlated directly with the levels of dietary folate ( r = 0.82, P < 0.001 and r = 0.35, P = 0.035, respectively) (Table I ). Colonic folate concentrations reached a plateau beyond four times the basal requirement of folate, which is related to the fact that folate accumulation in tissues is limited by the level of folylpolyglutamate synthetase activity in the setting of substrate excess ( 33 ) and is consistent with previous studies ( 4 , 34 ). Plasma and colonic folate concentrations in the negative control (i.e. non-DMH-treated rats receiving 2 mg folate/kg) were not significantly different from the corresponding values in the DMH-treated rats receiving 2 mg folate/kg. This indicates that the DMH treatment did not directly affect folate concentrations.
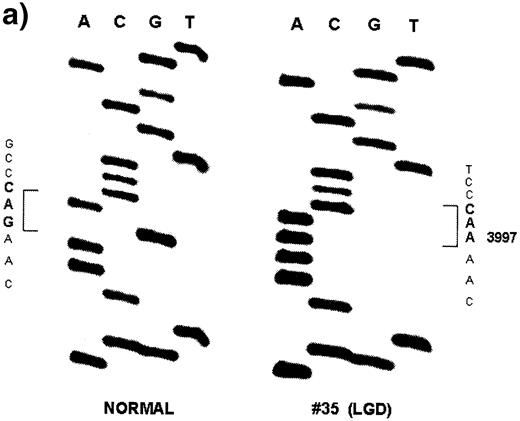
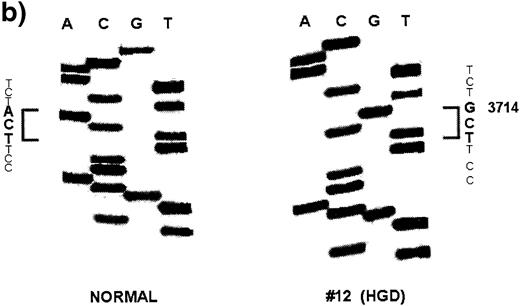
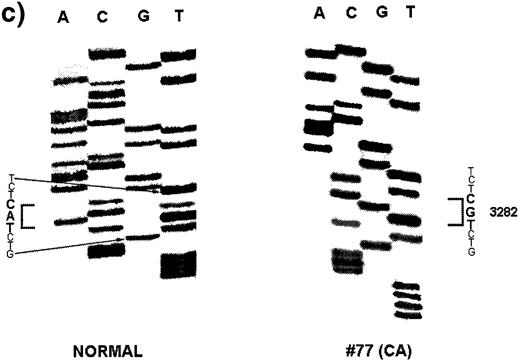
A total of six Apc mutations were found in four dysplastic lesions and invasive adenocarcinomas: two in two LGD, two in one LGD and two in one CA (Table II ). The combined mutation frequency of Apc in all the neoplastic lesions studied was 19.4% (six mutations out of 31 lesions). The frequency of mutation was not significantly different among the three neoplastic groups: 18.2% in LGD (two out of 11), 28.6% in HGD (two out of seven) and 15.4% in CA (two out of 13) (Table II ). No mutations were observed in 10 DMH-treated normal controls and five non-DMH-treated negative controls. A representative sequencing autoradiogram is shown in Figure 1 . The mutation frequency of Apc in the present study is comparable with the previously reported rate of 14.3% in colonic tumors induced by azoxymethane, a metabolite of DMH, in F344 rats ( 35 ). The Apc mutation frequency, however, varies from 0 to 100% in other chemical rodent models of CRC (29,36,37; Table III ). Discrepancies in the frequency of Apc mutations among these chemical rodent models are likely due to a species difference, the type, dose and duration of chemical carcinogens used, the sensitivity of mutation analyses and the extent of coding region examined. In human colon adenomas and carcinomas, the frequency of APC mutations varies from 40 to 80% ( 13 , 14 , 23 ). The relatively low frequency of Apc mutations observed in the present study compared with that observed in human CRC might be related to the fact that this study only analyzed Apc mutations within a 2704 bp region flanking the sequences corresponding to the human MCR. It is possible that an analysis of the entire coding region of the Apc gene (8526 bp) might have detected a higher frequency of mutations.
One unique finding in the present study is that all six Apc mutations were single base substitutions resulting in missense mutations (Table II ). Furthermore, two mutations in one HGD lesion were polymorphisms without an amino acid change (Table II ). Four of the six mutations (67%) were A:T→G:C transitions (Table II ). Some of the published animal studies ( 35 , 37 ) only screened for truncating mutations and, hence, the frequency of missense mutations could not be determined. Among the three studies which screened for all Apc mutations ( 29 , 36 ), only one missense mutation (a T:A→C:G transition at nt 1567) was identified ( 29 ). In human colon adenomas and carcinomas, >90% of somatic APC mutations are nonsense or frameshift mutations resulting in truncation of the APC protein ( 13 , 14 , 23 ). The frequency of the A:T→G:C transitional point mutation ranges from 3.5 to 10.9% in two published APC mutation databases for human CRC ( 13 , 14 ). Therefore, our finding of the predominant missense Apc point mutations, >2/3 of which are A:T→G:C transitions, may be unique to the DMH-induced colon tumors in Sprague–Dawley rats.
In contrast to observations made in human CRC, where >60% of the somatic APC mutations are clustered in the MCR in exon 15 ( 13 , 14 , 23 ), five of the six Apc mutations (83%) observed in the present study were located upstream (nt 3078–3714) from the MCR and only one mutation was found in the MCR (nt 3906–4589) (Table II ). This observation is consistent with the previous finding in azoxymethane-induced colon tumors in F344 rats ( 35 ). In the latter study, 100% of the observed Apc mutations were found upstream (nt 3173–3835) of the MCR ( 35 ). Also, in heterocyclic amine-induced colon tumors in F344 rats, only 28.6% of Apc mutations were observed in the MCR, whereas 28.6% were located between nt 2606 and 2761 in exon 15 and 42.8% in other exons ( 29 ). Thus, in DMH- or azoxymethane-induced colon tumors in rats, the MCR for human CRC is not commonly mutated and a 757 bp region between nt 3078 and 3835 in exon 15 upstream of the MCR may be a mutational hot spot. Because the entire coding sequence of the Apc gene was not scanned in the present study, the possibility of other mutational hot spots of the Apc gene for the DMH rat model of CRC cannot be ruled out.
In the present study, Apc mutations were found in 18.2% of the LGD lesions studied and the frequency of mutations did not significantly increase in HGD (28.6%) and CA (15.4%). This observation suggests that Apc mutations are an early molecular event in DMH-induced colorectal carcinogenesis in rats. This is consistent with human colorectal carcinogenesis, where APC mutations are considered to be an early event and a uniformly high mutation frequency of ~60% is observed in both colorectal adenomas and carcinomas ( 13 , 14 , 23 ).
Although Apc mutations were observed only in neoplastic lesions from rats receiving either 2 or 8 mg folate/kg diet (mutation frequency 10 and 21%, respectively; Table II ), the frequency of mutations among the four diet groups was not significantly different. Interestingly, no C:G→T:A transitional Apc mutations at CpG sites, which are expected from folate deficiency via its effects on DNA methylation and account for >50% of somatic APC mutations within the MCR in human CRC ( 13 , 14 , 23 ), were found in neoplastic lesions in the present study. Our data suggest that dietary folate had no significant effect on the frequency and type of Apc mutations within the 2704 bp region in exon 15 examined in the present study. However, we cannot exclude the possibility that the low frequency of Apc mutations and the small number of neoplasms studied in this study precluded our ability to observe modulatory effects of folate.
In contrast to the Apc gene, no mutations were observed in the mutational hot spots of the p53 gene for human colorectal cancer (exons 5–9) in all DMH-induced colorectal neoplasms, DMH-treated normal controls and non-DMH-treated negative controls. Most important of all was our observation that the superimposition of folate depletion, a condition known to induce p53 hypomethylation and strand breaks in rats ( 19 , 20 ), did not result in p53 mutations. Our observation is consistent with previous animal studies utilizing different carcinogens in different strains of rats or mice ( 38 – 45 ). Only one of these eight published studies demonstrated p53 mutations in exons 5–8 in DMH-treated mice with an overall mutation frequency of 11% ( 44 ), which is much lower than that observed in human CRC (>70%) ( 15 ). Taken together, these data suggest that p53 mutations are either absent or uncommonly involved in colorectal carcinogenesis in DMH or its metabolite, azoxymethane, rodent models. This fact implies that CRC in these rodent models may develop along a different pathway(s) independent of or by-passing the tumor suppressive function of the p53 gene commonly observed in human colorectal carcinogenesis.
In summary, the present study suggests that the Apc gene is mutated in a distinct region upstream of the human APC MCR in early stages of the DMH Sprague–Dawley rat model of CRC, albeit to a lesser degree than observed in human CRC. Unlike human and other rodent models of CRC, all mutations are single base missense mutations (mostly A:T→G:C transitions) not resulting in truncated Apc proteins. In contrast, the mutational hot spot of the p53 gene for human CRC is not mutated in this model. This study also suggests that the mechanism by which dietary folate modulates colorectal carcinogenesis in this model ( 4 ) is probably not via its effect on mutations in the mutational hot spots of the Apc and p53 genes.
Proportion of microscopic colorectal neoplasms and plasma and colonic mucosal folate concentrations
| Diet (mg folate/kg diet) | n | Histology | Plasma folate a (ng/ml) | Colon folate a (ng/g tissue) | |||
|---|---|---|---|---|---|---|---|
| Normal | LGD | HGD | CA | ||||
| a Mean ± SEM. | |||||||
| b–e Means with different superscript letters in each column are significantly different, P < 0.04 (Fisher's least significant difference multiple comparison test). | |||||||
| 0 | 8 | 4/8 | 2/8 | 0/8 | 2/8 | 16.69 ± 10.47 b | 388.94 ± 78.88 b |
| 2 | 10 | 3/10 | 2/10 | 2/10 | 3/10 | 55.42 ± 9.36 c | 629.86 ± 69.56 c |
| 8 | 14 | 2/14 | 4/14 | 4/14 | 4/14 | 91.54 ± 7.91 d | 929.05 ± 55.78 d |
| 40 | 9 | 1/9 | 3/9 | 1/9 | 4/9 | 164.61 ± 9.87 e | 861.17 ± 78.88 d |
| Diet (mg folate/kg diet) | n | Histology | Plasma folate a (ng/ml) | Colon folate a (ng/g tissue) | |||
|---|---|---|---|---|---|---|---|
| Normal | LGD | HGD | CA | ||||
| a Mean ± SEM. | |||||||
| b–e Means with different superscript letters in each column are significantly different, P < 0.04 (Fisher's least significant difference multiple comparison test). | |||||||
| 0 | 8 | 4/8 | 2/8 | 0/8 | 2/8 | 16.69 ± 10.47 b | 388.94 ± 78.88 b |
| 2 | 10 | 3/10 | 2/10 | 2/10 | 3/10 | 55.42 ± 9.36 c | 629.86 ± 69.56 c |
| 8 | 14 | 2/14 | 4/14 | 4/14 | 4/14 | 91.54 ± 7.91 d | 929.05 ± 55.78 d |
| 40 | 9 | 1/9 | 3/9 | 1/9 | 4/9 | 164.61 ± 9.87 e | 861.17 ± 78.88 d |
Proportion of microscopic colorectal neoplasms and plasma and colonic mucosal folate concentrations
| Diet (mg folate/kg diet) | n | Histology | Plasma folate a (ng/ml) | Colon folate a (ng/g tissue) | |||
|---|---|---|---|---|---|---|---|
| Normal | LGD | HGD | CA | ||||
| a Mean ± SEM. | |||||||
| b–e Means with different superscript letters in each column are significantly different, P < 0.04 (Fisher's least significant difference multiple comparison test). | |||||||
| 0 | 8 | 4/8 | 2/8 | 0/8 | 2/8 | 16.69 ± 10.47 b | 388.94 ± 78.88 b |
| 2 | 10 | 3/10 | 2/10 | 2/10 | 3/10 | 55.42 ± 9.36 c | 629.86 ± 69.56 c |
| 8 | 14 | 2/14 | 4/14 | 4/14 | 4/14 | 91.54 ± 7.91 d | 929.05 ± 55.78 d |
| 40 | 9 | 1/9 | 3/9 | 1/9 | 4/9 | 164.61 ± 9.87 e | 861.17 ± 78.88 d |
| Diet (mg folate/kg diet) | n | Histology | Plasma folate a (ng/ml) | Colon folate a (ng/g tissue) | |||
|---|---|---|---|---|---|---|---|
| Normal | LGD | HGD | CA | ||||
| a Mean ± SEM. | |||||||
| b–e Means with different superscript letters in each column are significantly different, P < 0.04 (Fisher's least significant difference multiple comparison test). | |||||||
| 0 | 8 | 4/8 | 2/8 | 0/8 | 2/8 | 16.69 ± 10.47 b | 388.94 ± 78.88 b |
| 2 | 10 | 3/10 | 2/10 | 2/10 | 3/10 | 55.42 ± 9.36 c | 629.86 ± 69.56 c |
| 8 | 14 | 2/14 | 4/14 | 4/14 | 4/14 | 91.54 ± 7.91 d | 929.05 ± 55.78 d |
| 40 | 9 | 1/9 | 3/9 | 1/9 | 4/9 | 164.61 ± 9.87 e | 861.17 ± 78.88 d |
Summary of the Apc mutations found in rat colon neoplastic lesions induced by DMH
| Rat | Diet (g folate/kg diet) | Histology | Nucleotide position a | Amino acid position | Nucleotide change | Result |
|---|---|---|---|---|---|---|
| a Nucleotide numbers are assigned according to the rat Apc cDNA sequence ( 40 ). | ||||||
| 35 | 8 | LGD | 3997 | 1333 | G AC→ A AC | Asp→Asn |
| 105 | 2 | LGD | 3623 | 1208 | A A G→A G G | Gln→Arg |
| 12 | 8 | HGD | 3714 | 1238 | TC A →TC G | Ser→Ser |
| 12 | 8 | HGD | 3078 | 1026 | AG T →AG A | Ser→Ser |
| 77 | 8 | CA | 3282 | 1094 | T A C→T G C | Tyr→Cys |
| 77 | 8 | CA | 3679 | 1227 | A AC→ G AC | Asn→Asp |
| Rat | Diet (g folate/kg diet) | Histology | Nucleotide position a | Amino acid position | Nucleotide change | Result |
|---|---|---|---|---|---|---|
| a Nucleotide numbers are assigned according to the rat Apc cDNA sequence ( 40 ). | ||||||
| 35 | 8 | LGD | 3997 | 1333 | G AC→ A AC | Asp→Asn |
| 105 | 2 | LGD | 3623 | 1208 | A A G→A G G | Gln→Arg |
| 12 | 8 | HGD | 3714 | 1238 | TC A →TC G | Ser→Ser |
| 12 | 8 | HGD | 3078 | 1026 | AG T →AG A | Ser→Ser |
| 77 | 8 | CA | 3282 | 1094 | T A C→T G C | Tyr→Cys |
| 77 | 8 | CA | 3679 | 1227 | A AC→ G AC | Asn→Asp |
Summary of the Apc mutations found in rat colon neoplastic lesions induced by DMH
| Rat | Diet (g folate/kg diet) | Histology | Nucleotide position a | Amino acid position | Nucleotide change | Result |
|---|---|---|---|---|---|---|
| a Nucleotide numbers are assigned according to the rat Apc cDNA sequence ( 40 ). | ||||||
| 35 | 8 | LGD | 3997 | 1333 | G AC→ A AC | Asp→Asn |
| 105 | 2 | LGD | 3623 | 1208 | A A G→A G G | Gln→Arg |
| 12 | 8 | HGD | 3714 | 1238 | TC A →TC G | Ser→Ser |
| 12 | 8 | HGD | 3078 | 1026 | AG T →AG A | Ser→Ser |
| 77 | 8 | CA | 3282 | 1094 | T A C→T G C | Tyr→Cys |
| 77 | 8 | CA | 3679 | 1227 | A AC→ G AC | Asn→Asp |
| Rat | Diet (g folate/kg diet) | Histology | Nucleotide position a | Amino acid position | Nucleotide change | Result |
|---|---|---|---|---|---|---|
| a Nucleotide numbers are assigned according to the rat Apc cDNA sequence ( 40 ). | ||||||
| 35 | 8 | LGD | 3997 | 1333 | G AC→ A AC | Asp→Asn |
| 105 | 2 | LGD | 3623 | 1208 | A A G→A G G | Gln→Arg |
| 12 | 8 | HGD | 3714 | 1238 | TC A →TC G | Ser→Ser |
| 12 | 8 | HGD | 3078 | 1026 | AG T →AG A | Ser→Ser |
| 77 | 8 | CA | 3282 | 1094 | T A C→T G C | Tyr→Cys |
| 77 | 8 | CA | 3679 | 1227 | A AC→ G AC | Asn→Asp |
Summary of Apc mutations in rodent models of colorectal cancer
| Reference | Species | Diagnosis ( n ) | Carcinogen | Exons examined | Mutation analysis | Outcome |
|---|---|---|---|---|---|---|
| a AOM, azoxymethane; DMH, dimethylhydrazine; HA, 1-hydroxyanthraquinone; IQ, 2-amino-3-methylimidazo(4,5- f )quinoline; MAM, methylazoxymethanol; PhIP, 2-amino-1-methyl-6-phenylimidazo(4,5- b )pyridine. | ||||||
| b PCR–SSCP, PCR–single strand conformation polymorphism; IVSP, In vitro synthesized protein assay. | ||||||
| 29 | Male F344 Rats | CA (8) | IQ a | 1–15 | PCR–SSCP b | Mutation frequency, 15.4% |
| Direct sequencing | 1 missense mutation | |||||
| 1 truncating mutation | ||||||
| 29 | Male F344 rats | CA (11) & adenoma (2) | PhIP | 1–15 | PCR–SSCP | Mutation frequency, 62.5% |
| Direct sequencing | All truncating mutations | |||||
| 35 | Male F344 rats | CA (10), adenoma (17) & unknown (1) | AOM | 15 (nt 2131–4750) | IVSP | Mutation frequency, 14.3% |
| All truncating mutations | ||||||
| 36 | Male F344 rats | CA (16) & adenoma (4) | HA & MAM | 1–15 | PCR–SSCP | No mutations |
| 37 | CF-1 mice | CA (3) & adenoma (26) | AOM | 1–15 | Immunohistochemistry | Mutation frequency, 100% |
| All truncating mutations | ||||||
| Present study | Male Sprague–Dawley rats | Normal (10), LGD (11), HGD (7) & CA (13) | DMH | 15 (nt 2074–4778) | Direct sequencing | Mutation frequency, 19.4% |
| All missense mutations | ||||||
| Reference | Species | Diagnosis ( n ) | Carcinogen | Exons examined | Mutation analysis | Outcome |
|---|---|---|---|---|---|---|
| a AOM, azoxymethane; DMH, dimethylhydrazine; HA, 1-hydroxyanthraquinone; IQ, 2-amino-3-methylimidazo(4,5- f )quinoline; MAM, methylazoxymethanol; PhIP, 2-amino-1-methyl-6-phenylimidazo(4,5- b )pyridine. | ||||||
| b PCR–SSCP, PCR–single strand conformation polymorphism; IVSP, In vitro synthesized protein assay. | ||||||
| 29 | Male F344 Rats | CA (8) | IQ a | 1–15 | PCR–SSCP b | Mutation frequency, 15.4% |
| Direct sequencing | 1 missense mutation | |||||
| 1 truncating mutation | ||||||
| 29 | Male F344 rats | CA (11) & adenoma (2) | PhIP | 1–15 | PCR–SSCP | Mutation frequency, 62.5% |
| Direct sequencing | All truncating mutations | |||||
| 35 | Male F344 rats | CA (10), adenoma (17) & unknown (1) | AOM | 15 (nt 2131–4750) | IVSP | Mutation frequency, 14.3% |
| All truncating mutations | ||||||
| 36 | Male F344 rats | CA (16) & adenoma (4) | HA & MAM | 1–15 | PCR–SSCP | No mutations |
| 37 | CF-1 mice | CA (3) & adenoma (26) | AOM | 1–15 | Immunohistochemistry | Mutation frequency, 100% |
| All truncating mutations | ||||||
| Present study | Male Sprague–Dawley rats | Normal (10), LGD (11), HGD (7) & CA (13) | DMH | 15 (nt 2074–4778) | Direct sequencing | Mutation frequency, 19.4% |
| All missense mutations | ||||||
Summary of Apc mutations in rodent models of colorectal cancer
| Reference | Species | Diagnosis ( n ) | Carcinogen | Exons examined | Mutation analysis | Outcome |
|---|---|---|---|---|---|---|
| a AOM, azoxymethane; DMH, dimethylhydrazine; HA, 1-hydroxyanthraquinone; IQ, 2-amino-3-methylimidazo(4,5- f )quinoline; MAM, methylazoxymethanol; PhIP, 2-amino-1-methyl-6-phenylimidazo(4,5- b )pyridine. | ||||||
| b PCR–SSCP, PCR–single strand conformation polymorphism; IVSP, In vitro synthesized protein assay. | ||||||
| 29 | Male F344 Rats | CA (8) | IQ a | 1–15 | PCR–SSCP b | Mutation frequency, 15.4% |
| Direct sequencing | 1 missense mutation | |||||
| 1 truncating mutation | ||||||
| 29 | Male F344 rats | CA (11) & adenoma (2) | PhIP | 1–15 | PCR–SSCP | Mutation frequency, 62.5% |
| Direct sequencing | All truncating mutations | |||||
| 35 | Male F344 rats | CA (10), adenoma (17) & unknown (1) | AOM | 15 (nt 2131–4750) | IVSP | Mutation frequency, 14.3% |
| All truncating mutations | ||||||
| 36 | Male F344 rats | CA (16) & adenoma (4) | HA & MAM | 1–15 | PCR–SSCP | No mutations |
| 37 | CF-1 mice | CA (3) & adenoma (26) | AOM | 1–15 | Immunohistochemistry | Mutation frequency, 100% |
| All truncating mutations | ||||||
| Present study | Male Sprague–Dawley rats | Normal (10), LGD (11), HGD (7) & CA (13) | DMH | 15 (nt 2074–4778) | Direct sequencing | Mutation frequency, 19.4% |
| All missense mutations | ||||||
| Reference | Species | Diagnosis ( n ) | Carcinogen | Exons examined | Mutation analysis | Outcome |
|---|---|---|---|---|---|---|
| a AOM, azoxymethane; DMH, dimethylhydrazine; HA, 1-hydroxyanthraquinone; IQ, 2-amino-3-methylimidazo(4,5- f )quinoline; MAM, methylazoxymethanol; PhIP, 2-amino-1-methyl-6-phenylimidazo(4,5- b )pyridine. | ||||||
| b PCR–SSCP, PCR–single strand conformation polymorphism; IVSP, In vitro synthesized protein assay. | ||||||
| 29 | Male F344 Rats | CA (8) | IQ a | 1–15 | PCR–SSCP b | Mutation frequency, 15.4% |
| Direct sequencing | 1 missense mutation | |||||
| 1 truncating mutation | ||||||
| 29 | Male F344 rats | CA (11) & adenoma (2) | PhIP | 1–15 | PCR–SSCP | Mutation frequency, 62.5% |
| Direct sequencing | All truncating mutations | |||||
| 35 | Male F344 rats | CA (10), adenoma (17) & unknown (1) | AOM | 15 (nt 2131–4750) | IVSP | Mutation frequency, 14.3% |
| All truncating mutations | ||||||
| 36 | Male F344 rats | CA (16) & adenoma (4) | HA & MAM | 1–15 | PCR–SSCP | No mutations |
| 37 | CF-1 mice | CA (3) & adenoma (26) | AOM | 1–15 | Immunohistochemistry | Mutation frequency, 100% |
| All truncating mutations | ||||||
| Present study | Male Sprague–Dawley rats | Normal (10), LGD (11), HGD (7) & CA (13) | DMH | 15 (nt 2074–4778) | Direct sequencing | Mutation frequency, 19.4% |
| All missense mutations | ||||||
A representative sequencing autoradiogram for rat Apc mutations from ( a ) low grade dysplasia (LGD), ( b ) high grade dysplasia (HGD) and ( c ) invasive adenocarcinoma (CA).
To whom correspondence should be addressed Email: youngin.kim@utoronto.ca
Presented in part at the 1998 Digestive Disease Week, May 17–20, 1998, New Orleans, LA, USA, and published in abstract form in Gastroenterology , 1998, 114, G2571.
We thank the animal caretakers of the Jean Mayer USDA Human Nutrition Research Center on Aging at Tufts University for feeding and maintenance of the rats used in this study. We also thank Helixx Technologies Inc. for technical support. This project has been supported in part by grants from the National Cancer Institute (1-UO1 CA63812-01 to J.B.M.), the American Institute for Cancer Research (J.B.M.), the US Department of Agriculture, Agricultural Research Service Contract 53-3K06-01 (J.B.M.), an Operating Grant, Fellowship and Scholarship from the Medical Research Council of Canada (Y.-I.K.), a Future Leader Award from the North American Branch of the International Life Sciences Institute (Y.-I.K.) and a St Michael's Hospital Health Sciences Research Program Summer Student Scholarship (M.P.).
References
Kim,Y.-I. (
Mason,J.B. and Levesque,T. (
Cravo,M.L., Mason,J.B., Dayal,Y., Hutchinson,M., Smith,D., Selhub,J. and Rosenberg,I.H. (
Kim,Y.-I., Salomon,R.N., Graeme-Cook,F., Choi,S.-W., Smith,D.E., Dallal,G.E. and Mason,J.B. (
Wagner,C. (1995) Biochemical role of folate in cellular metabolism. In Bailey,L.B. (ed.) Folate in Health and Disease . Marcel Dekker, New York, NY, pp. 23–42.
Haynes,R.H. (1985) Molecular mechanisms in genetic stability and change: the role of deoxyribonucleotide pool balance. In de Serres,F.S. (ed.) Genetic Consequences of Nucleotide Pool Imbalance . Plenum, New York, NY, pp. 1–23.
James,S.J., Cross,D.R. and Miller,B.J. (
James,S.J., Basnakian,A.G. and Miller,B.J. (
James,S.J., Miller,B.J., Basnakian,A.G., Pogribny,I.P., Pogribna,M. and Muskhelishvili,L. (
Kunz,B.A. and Kohalmi,S.E. (
Zingg,J.-M. and Jones,P.A. (
Laird,P.W. and Jaenisch,R. (
Nagase,H. and Nakamura,Y. (
Beroud,C. and Soussi,T. (
Hollstein,M., Sidransky,D., Vogelstein,B. and Harris,C.C. (
Rideout,W.M., Coetzee,G.A., Olumi,A.F. and Jones,P.A. (
Shen,J.C., Rideout,W.M. and Jones,P.A. (
Chen,R.Z., Pettersson,U., Beard,C., Jackson-Grusby,L. and Jaenisch,R. (
Kim,Y.-I., Pogribny,I.P., Basnakian,A.G., Miller,J.W., Selhub,J., James,S.J. and Mason,J.B. (
Kim,Y.-I., Pogribny,I.P., Salomon,R.N., Choi,S.W., Smith,D.E., James,S,J. and Mason,J.B. (
Branda,R.F. and Blickensderfe,D.B. (
Branda,R.F., Lafayette,A.R., O'Neill,J.P. and Nicklas,J.A. (
Fearon,E.R. and Vogelstein,B. (
Walzem,R.L. and Clifford,A.J. (
Filipe,M.I. (
Riddell,R.H., Goldman,H., Ransohoff,D.F., Appelman,H.D., Fenoglio, C.M., Haggitt,R.C., Ahren,C., Correa,P., Hamilton,S.R., Morson,B.C., Sommers,S.C. and Yardley,J.H. (
Tamura,T. (1990) Microbiological assay of folate. In Picciano,M.F., Stokstad,E.L.R. and Gregory,J.F. (eds) Folic Acid Metabolism in Health and Disease . Wiley-Liss, New York, NY, pp. 121–137.
Reitmair,A.H., Schmits,R., Ewel,A., Bapat,B., Redston,M., Mitri,A.S., Waterhouse,P., Mittrücker,H.-W., Wakeham,A., Liu,B., Thomason,A., Griesser,H., Gallinger,S., Ballhause,W.G., Fishel,R. and Mak,T.W. (
Kakiuchi,H., Watanabe,M., Ushijima,T., Toyota,M., Imai,K., Weisburger, J.H., Sugimura,T. and Nagao,M. (
Vancutsem,P.M., Lazarus,P. and Williams,G.M. (
Hulla,J.E. and Schneider,R.P. (
Anderson,R.D., Bao,C.Y., Minnick,D.T., Baxter,J., Veigl,M.L. and Sedwick,W.D. (
Shane,B. (1995) Folate chemistry and metabolism. In Bailey,L.B. (ed.) Folate in Health and Disease . Marcel Dekker, New York, NY, pp. 1–22.
Lowe,K.E., Osborne,C.B., Lin,B.-F., Kim,J.-S., Hus,J.-C. and Shane,B. (
De Filippo,C., Caderni,G., Bazzicalupo,M., Briani,C., Giannini,A., Fazi,M. and Dolara,P. (
Suzui,M., Ushijima,T., Yoshimi,N., Nakagama,H., Hara,A., Sugimura,T., Nagao,M. and Mori,H. (
Maltzman,T., Whittington,J., Driggers,L., Stephens,J. and Ahnen,D. (
Makino,H., Ushijima,U., Kakiuchi,H., Onda,M., Ito,N., Sugimura,T. and Nagao,M. (
Suzui,M., Yoshimi,N., Ushijima,T., Hirose,Y., Makita,H., Wang,A., Kawamori,T., Tanaka,T., Mori,H. and Nagao,M. (
Shivapurkar,N., Belinsky,S.A., Wolf,D.C., Tan,Z. and Alabaster,O. (
Shivapurkar,N., Nikula,N.J., Tanaka,T., Tang,Z.C. and Alabaster,O. (
Erdman,S.H., Wu,H.D., Hixson,L.J., Ahnen,D.J. and Gerner,E.W. (
Okamoto,M., Ohtsu,H., Miyaki,M. and Yonekawa,H. (
Okamoto,M., Ohtsu,H., Kominami,R. and Yonekawa,H. (
Smits,R., Kartheuser,A., Jagmohan-Changur,S., Leblanc,V., Breukel,C., de Vries,A., van Kranen,H., van Krieke,H.H., Williamson,S., Edelman,W., Kucherlapati,R., Khan,P.M. and Fodde,R. (

{kind=link}