




Abstract
Certain hexavalent chromium [Cr(VI)] compounds are human lung carcinogens. Although much is known about Cr-induced DNA damage, very little is known about mechanisms of Cr(VI) mutagenesis and the role that DNA repair plays in this process. Our goal was to investigate the role of excision repair (ER) pathways in Cr(VI)-mediated mutagenesis in mammalian cells. Repair-proficient Chinese hamster ovary cells (AA8), nucleotide excision repair (NER)-deficient (UV-5) and base excision repair (BER)-inhibited cells were treated with Cr(VI) and monitored for forward mutation frequency at the hypoxanthine-guanine phosphoribosyltransferase (HPRT) locus. BER was inhibited using methoxyamine hydrochloride (Mx), which binds to apurinic/apyrimidinic sites generated during BER. Notably, we found that both NER-deficient (UV-5 and UV-41) and BER-inhibited (AA8 + Mx) cells displayed attenuated Cr(VI) mutagenesis. To determine whether this was unique to Cr(VI), we included the alkylating agent, methylmethane sulfonate (MMS) and ultraviolet (UV) radiation (260 nm) in our studies. Similar to Cr(VI), UV-5 cells exhibited a marked attenuation of MMS mutagenesis, but were hypermutagenic following UV exposure. Moreover, UV-5 cells expressing human xeroderma pigmentosum complementation group D displayed similar sensitivity to Cr(VI) and MMS-induced mutagenesis as AA8 controls, indicating that the genetic loss of NER was responsible for attenuated mutagenesis. Interestingly, Cr(VI)-induced clastogenesis was also attenuated in NER-deficient and BER-inhibited cells. Taken together, our results suggest that NER and BER are required for Cr(VI) and MMS-induced genomic instability. We postulate that, in the absence of ER, DNA damage is channeled into an error-free system of DNA repair or damage tolerance.
Introduction
Certain chromium containing compounds are classified by the International Agency for Cancer Research as group I human lung carcinogens, as they are encountered in certain occupational settings. Generally, these are moderately insoluble particulates that dissolve slowly after tissue deposition and cell-surface adherence, releasing soluble hexavalent chromium [Cr(VI)] directly onto or into nearby cells ( 1 ). Soluble Cr(VI) exists as an oxyanion that is isostructural with sulfate and enters the cell via sulfate anion transporters ( 1 ). The Cr(VI) oxyanion is itself unreactive but is intracellularly reduced to trivalent chromium [Cr(III)], the most stable valence state of Cr, which has an affinity for macromolecules including DNA ( 2 ) and proteins ( 3 , 4 ). The inability of Cr(III) to cross the plasma membrane allows for millimolar levels of Cr(III) to accumulate intracellularly ( 5 , 6 ). Another reactive, but less stable, valence state generated by Cr(VI) reduction is Cr(V) ( 7 ). The intracellular reduction of Cr(VI) produces a broad spectrum of DNA lesions including binary DNA adducts, DNA interstrand cross-links (ICLs), DNA–protein adducts, DNA double-strand breaks and oxidized bases ( 1 , 2 , 8–13 ).
Nucleotide excision repair (NER) is a versatile repair mechanism responsible for the removal of large helix-distorting lesions such as bulky DNA adducts and ICLs (reviewed in ref. 14 and refs 15 , 16 , respectively). In humans, the NER pathway was initially elucidated in patients afflicted with xeroderma pigmentosum (XP). Loss of a single XP gene inactivates the entire NER pathway, as there are not any functionally redundant genes in NER ( 17 ). It has been published previously by our laboratory and others that NER plays a pivotal role in the survival of mammalian cells after Cr(VI) exposure ( 18 , 19 ). For example, the loss of either xeroderma pigmentosum complementation group D (XPD) (5′ to 3′ helicase) or xeroderma pigmentosum complementation group F (XPF) (5′-endonuclease) increases the sensitivity toward Cr(VI) lethality and impairs the removal of Cr–DNA adducts ( 18 ). Moreover, we have also shown that Cr–DNA damage is a substrate for prokaryotic UvrABC endonuclease, the primary NER complex for the removal of bulky lesions in prokaryotes ( 20 ).
Cr(VI) is a complex genotoxicant. Accordingly, several lesions generated by Cr(VI) reduction (i.e. oxidized bases) are substrates for base excision repair (BER). In BER, damaged (alkylated or oxidized) bases are recognized by specific DNA glycosylases and are excised, resulting in the formation of an apurinic/apyrimidinic (AP) site. AP endonucleases (i.e. APN1/2 in yeast and APE1 in humans) recognize AP sites and cleave the phosphodiester backbone in nicked DNA, which is then filled and ligated by DNA polymerase β and DNA ligase IV, respectively ( 21 ). It has been suggested that the transient intermediate, Cr(V), can facilitate the oxidation of existing 8-oxoguanine lesions to form the premutagenic precursors guanidinohydantoin and spiroiminodihydantoin lesions ( 22 , 23 ). Guanidinohydantoin and spiroiminodihydantoin lesions are more potent polymerase-arresting lesions than 8-oxoguanine and are recognized by the NEIL glycosylases of the BER pathway ( 7 ).
Very little is known regarding the mechanism of Cr(VI) mutagenesis. NER plays a significant role in the removal of Cr-damaged DNA ( 18–20 ), but it is not known what, if any role BER plays in protecting cells from Cr(VI) toxicity and mutagenesis. In order to elucidate the relative contribution of these excision repair (ER) pathways, we have studied Cr(VI) mutagenesis in both NER-deficient Chinese hamster ovary (CHO) cells (UV-5 and UV-41) and CHO cells in which BER was inactivated by the pharmacological agent methoxyamine hydrochloride (Mx). Although we anticipated a dramatic increase in mutagenesis in ER-deficient cells, we actually observed a marked attenuation of Cr(VI)-induced mutagenesis in the absence of NER or BER. The attenuation of mutagenesis was observed in both NER-deficient cell lines, indicating that the effect was not unique to a particular NER gene. In addition, we found that methylmethane sulfonate (MMS)-induced mutagenesis was similarly attenuated by ER deficiency, indicating that this effect was not exclusive to Cr(VI). As reported previously, the NER-deficient strains were hypermutagenic to ultraviolet (UV) light, validating that our experimental system was capable of detecting increased mutagenesis. Our results collectively suggest that NER and BER are required for chemical mutagenesis and indicate that, in the absence of DNA ER, DNA base damage may be channeled into another more error-free repair pathway.
Materials and methods
Cell culture and drug preparation
Parental CHO cells (AA8, wild-type (WT)), XPD (UV-5) and XPF (UV-41) cells were maintained in minimal Eagle’s medium (MEM) (Invitrogen Life Technologies, Gaithersburg, MD), supplemented with 10% fetal bovine serum (Inovar, Gaithersburg, MD), 1% penicillin–streptomycin (Gibco, Grand Island, NY) and 2 mM glutamine (Fisher Scientific, Pittsburgh, PA) in a 95% air and 5% CO 2 humidified atmosphere at 37°C. WT-7 (AA8 expressing human XPD) and UV-5 (expressing human XPD) (kind gift from Dr Larry Thompson) were maintained in a medium containing 0.5 mg/ml G418 at all times. Cr(VI) [Na 2 CrO 4 •4H 2 O] (J.T. Baker Chemical, Phillipsburg, NJ) was freshly prepared in sterile, distilled, deionized water before each experiment. MMS was diluted in MEM prior to each experiment. Mx (Sigma, St Louis, MO) was used at a concentration of 0.5 mg/ml in complete MEM. MMS (Acros Organics, Geel, Belgium) was diluted from a stock concentration in dimethyl sulfoxide prior to each experiment.
Clonogenic survival and dosing regimens
Cultures were seeded (1 × 10 5 cells per plate) and allowed to adhere for 24 h. Cells were incubated with the indicated concentrations of Cr(VI) (24 h) or MMS (1 h) in complete culture medium. Cultures were washed with 1× PBS and recovered in complete culture medium for an additional 24 h and then collected by trypsinization. Approximately 400 cells were seeded onto 60 mm dishes in duplicate and grown for 5–7 days until colonies became visible. Colonies (>10 cells) were stained, counted and percent survival was determined for each treatment relative to its vehicle control. Additionally, when indicated, medium was supplemented with Mx at 0.5 mg/ml during the appropriate dosing time and the subsequent 24 h recovery ( 24 ).
For experiments utilizing UV radiation, cultures were seeded (1 × 10 5 cells per plate) and allowed to adhere overnight. At 24 h, cultures were washed two times with PBS to remove residual MEM prior to UV dosing. Adherent cells were dosed using a 260 nm half-band width ±2.5 filter at ∼0.1 J/m 2 /s to the desired dose of 0.74 J/m 2 . Immediately following UV exposure, cells were trypsinized and plated for clonogenic survival.
HPRT mutagenesis
CHO cells were treated as described for clonogenic survival assays and ∼1 × 10 5 cells were seeded into a 150 mm capped culture flask and recovered for 5–7 days. Following recovery, ∼1 × 10 6 cells were seeded in 150 mm capped culture flasks and grown in complete medium supplemented with 60 μM 6-thioguanine in duplicate. Vented capped culture flasks were grown for 5–7 days until colonies were visible. Colonies were stained and counted. In order to account for plating efficiency, ∼400 cells were seeded in 60 mm dishes and placed in an incubator for 5–7 days until colonies became visible. Colonies were stained and counted, and plating efficiency was determined for each sample relative to vehicle control.
Clastogenicity
Chromosome damage was assessed as 100 metaphases per data point with background damage levels subtracted as described previously ( 25 ). Briefly, 100 000 cells were seeded in 60 mm dishes and allowed to grow for 24 h allowing cells to resume log phase growth. Cultures were then treated for 24 h. One hour prior to harvest, the dishes were treated with 2% colchicine. During the harvest, treatment medium was removed, and cells were washed with 1× phosphate-buffered saline and removed from the dish with 0.25% Trypsin–ethylenediaminetetraacetic acid 1×. Cells were centrifuged, the supernatant was removed, the pellet was resuspended and 10 ml of 0.075 M KCl (hypotonic solution) was added for 10 min. Afterward, 1 ml of methanol:acetic acid fixative (3:1) was added. The cells were centrifuged again, supernatant was removed and 10 ml of fixative was added for 20 min. Cells were centrifuged and fixative was changed two more times. Lastly, cells were dropped onto a clean wet and stained using a 5% Giemsa stain in Gurr’s buffer. One hundred metaphases per data point were analyzed in each experiment, and each experiment was repeated at least three times ( 26 ). Total damage was determined by the number of chromatid lesions, chromatid exchanges, rings, dicentrics, double minutes and acentric fragments present under each condition. The data are expressed as the number of chromosome aberrations per 100 metaphases (background subtracted).
Results
Cr(VI) and MMS mutagenesis is attenuated in NER-deficient CHO cells
The historical dogma regarding DNA repair and mutagenesis suggests that, in the absence of repair, premutagenic precursors will persist on DNA thereby contributing to increased mutagenesis. As such, we hypothesized that following treatment of ER-deficient CHO cells with Cr(VI), there would be a significant increase in mutagenesis at the HPRT locus. Initially, AA8 CHO cells were treated with Cr(VI) (1.5 or 6 μM), which resulted in a 2- and 3.4-fold increase in HPRT mutagenesis, respectively ( Figure 1A ). Surprisingly, NER-deficient UV-5 cells, at the same concentrations of Cr(VI), exhibited attenuation in Cr(VI) mutagenesis. Specifically, Cr(VI) mutagenesis at 1.5 and 6 μM Cr(VI) was reduced by 37 and 66%, respectively. To determine whether the results were limited to only XPD deficiency, we also studied XPF-deficient (UV-41) CHO cells. Similar to XPD cells, XPF deficiency also hyposensitized cells to Cr(VI) mutagenesis, reducing the mutagenesis produced by 1.5 and 6 μM Cr(VI) by 52 and 52%, respectively. Background mutation frequencies for AA8, UV-5 and UV-41 CHO cells were 1.78 × 10 −5 , 5.24 × 10 −5 and 3.3 × 10 −5 , respectively. It is noteworthy that Cr–DNA adduct levels were similar in AA8, UV-5 and UV-41 cells immediately following Cr(VI) exposure, indicating that differential uptake was not responsible for the observed attenuated mutagenesis in NER-deficient cells ( 18 ).
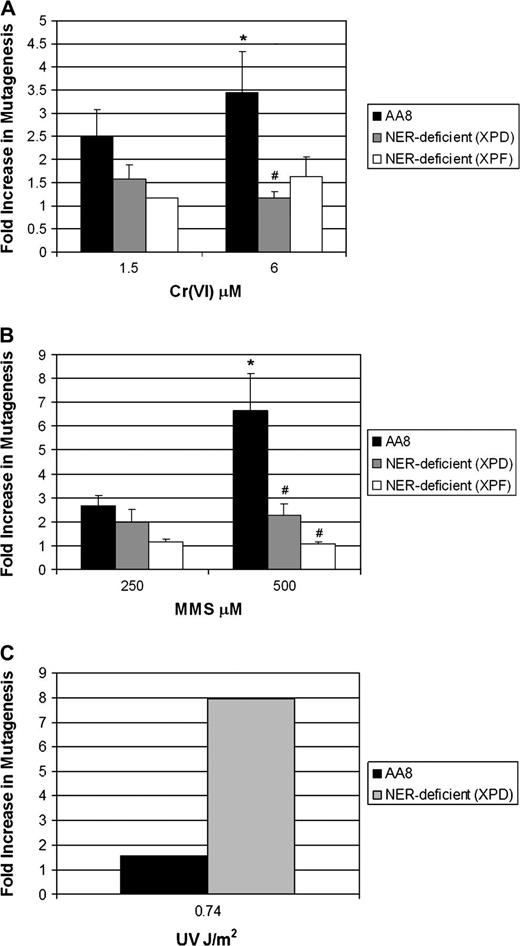
Loss of DNA ER attenuates Cr(VI) and MMS forward mutagenesis in mammalian cells. Repair-proficient (AA8) and repair-deficient (XPD and XPF) cells were treated with ( A ) Cr(VI) (as Na 2 CrO 4 ) for 24 h or ( B ) MMS for 1 h or ( C ) UV radiation (0.74 J/m 2 ); washed; plated in MEM (clonogenic survival) and seeded in MEM for recovery (5–7 days). Recovered cells were washed and seeded in MEM (plating efficiency) and MEM containing 6-thioguanine (mutant selection) for 5–7 days. The data are mean ± SD/SE of at least two (C) or three (A and B) independent experiments. *A significant increase in AA8 cells versus untreated control at the specified concentration as tested for by analysis of variance. #A statistical difference between AA8 and XPD or XPF at the specified concentration.
To determine if this result was specific to Cr(VI), we expanded our study to include the well-studied alkylating agent, MMS. MMS is highly mutagenic, predominantly generating 7-methylguanine and 3-methyladenine, which are both substrates for BER. However, it has been suggested that NER can serve as an alternative pathway in repairing methylated DNA bases ( 27 ). MMS treatment of AA8 CHO cells resulted in a concentration-dependent increase in HPRT mutagenesis (∼2- to 6.5-fold) at 250 and 500 μM, respectively. Consistent with our Cr(VI) data ( Figure 1A ), MMS-induced mutagenesis was attenuated by 26–84% in UV-5 and UV-41 cells compared with parental AA8 cells ( Figure 1B ).
It is well known that NER deficiency predisposes cells to increased UV-induced mutagenesis ( 17 , 28–31 ). To determine whether our NER-deficient cells were generally resistant to all mutagenesis, we measured HPRT mutation frequency following UV exposure. UV pyrimidine dimers are well-known substrates for NER and are highly mutagenic lesions ( 17 , 32 ). Consistent with previous work ( 18 ), repair-proficient, parental AA8 cells exhibited modest sensitivity to UV-induced lethality and mutagenesis. In contrast to the attenuated mutagenesis we observed with Cr(VI) and MMS, UV-5 and UV-41 cells were highly sensitive to UV lethality and were markedly hypermutagenic following UV exposure at 0.74 J/m 2 ( Figure 1C ).
NER is required for Cr(VI) and MMS mutagenesis
We have reported previously that UV-5 cells are impeded in the removal of Cr–DNA adducts ( 18 ). Moreover, UV-5 cells, complemented with human XPD, repaired Cr–DNA adducts comparable with parental AA8 cells. In order to determine whether the loss of NER was truly responsible for attenuated Cr(VI) and MMS mutagenesis, we treated UV-5 cells expressing human XPD with peak mutagenic concentrations of Cr(VI) (6 μM) or MMS (500 μM) and measured HPRT mutation frequency. XPD complemented AA8 and UV-5 CHO cells expressed background mutation frequencies of 5.35 × 10 −6 and 2.9 × 10 −5 , respectively. Interestingly, XPD expressing UV-5 cells displayed a sensitivity toward Cr(VI)- ( Figure 2A ) and MMS-induced ( Figure 2B ) mutagenesis that was similar to AA8 cells.
![NER is required for Cr(VI) and MMS mutagenesis in mammalian cells. Repair-proficient [AA8, AA8 (+XPD complelmentary DNA)], repair-deficient (XPD) and repair-complemented [XPD (+XPD complelmentary DNA)] cells were treated with ( A ) Cr(VI) for 24 h and ( B ) MMS for 1 h and plated in MEM (clonogenic survival) and seeded in MEM for recovery (5–7 days). Recovered cells were washed and seeded in MEM (plating efficiency) and MEM containing 6-thioguanine (mutant selection) for 5–7 days. The data are mean ± SE of at least three independent experiments. *A significant increase in AA8 and AA8 (+XPD) at the specified concentrations versus untreated controls as tested for by analysis of variance. #Significant differences between AA8 and AA8 (+XPD) and XPD cells at specified concentrations. +A significant difference between AA8 (+XPD) and XPD-deficient cells.](https://oup.silverchair-cdn.com/oup/backfile/Content_public/Journal/carcin/29/5/10.1093_carcin_bgn058/3/m_carcinbgn058f02_lw.jpeg?Expires=1716517708&Signature=jKP-zjEIqStF91cv2xRmc2az8f1xzcPbrrGZXwoGuLr6LDYovi6pMG3nsboD3t0cOpz1Xliv6NKjirv-PsWfNlMqnVluMaTAwUYFS2c8uIM7Zc~lJmIYEl6W8IqrtWFQaw~vtcUnPAWf9ox7RFdc5P1~6USlpA9H5A2ti4a94HsPDJf777dCoyWmGxf8Phq06Lx4XGLAKYiFU~VslGNYUOQwMHj-3ESCEC-XRlHxEPA-JcEppp0XXp0Twjso0Pf3Usq4y2ZEVjkzsFjT0jqNNNtArgFnBH2LCZDmNGP-YW8~azXlegS1hiIiN5aT02eMH9HsMUl-K7vahJ4oueQHQw__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
NER is required for Cr(VI) and MMS mutagenesis in mammalian cells. Repair-proficient [AA8, AA8 (+XPD complelmentary DNA)], repair-deficient (XPD) and repair-complemented [XPD (+XPD complelmentary DNA)] cells were treated with ( A ) Cr(VI) for 24 h and ( B ) MMS for 1 h and plated in MEM (clonogenic survival) and seeded in MEM for recovery (5–7 days). Recovered cells were washed and seeded in MEM (plating efficiency) and MEM containing 6-thioguanine (mutant selection) for 5–7 days. The data are mean ± SE of at least three independent experiments. *A significant increase in AA8 and AA8 (+XPD) at the specified concentrations versus untreated controls as tested for by analysis of variance. #Significant differences between AA8 and AA8 (+XPD) and XPD cells at specified concentrations. +A significant difference between AA8 (+XPD) and XPD-deficient cells.
BER plays a role distinct from NER in protecting CHO cells from Cr(VI) toxicity
The common intermediate step in BER involves the cleavage of AP sites generated as a consequence of DNA glyosylase-mediated excision of the damaged base. Unfortunately, there are no AP endonuclease-deficient CHO cells available to study the role of this enzyme in protecting cells from Cr(VI) toxicity and mutagenesis. Therefore, we pharmacologically inhibited BER in CHO cells using Mx and determined the impact that this had on Cr(VI) toxicity and mutageneis ( 24 , 33–35 ). Mx binds to the aldehyde group of AP sites, thereby preventing further processing by AP endonucleases. Several studies have demonstrated that Mx is an effective inhibitor of BER and potentiates cell killing by DNA base-damaging agents ( 24 , 34 ) and antimetabolites ( 33 ). As expected, we initially found that Mx significantly hypersensitized AA8 cells to hydrogen peroxide toxicity, a known DNA-oxidizing agent (data not shown). Using Mx, we investigated the potential role of BER in protecting CHO cells from Cr(VI) toxicity. AA8- and NER-deficient (UV-5) CHO cells were exposed to Cr(VI) (1.5 and 6 μM) in the presence or absence of Mx (0.5 mg/ml) for 24 h. Cultures were then washed and allowed to recover for 24 h in the appropriate media (±Mx) before being seeded for clonogenic survival. As shown in Figure 3 , BER-inhibited AA8 cells (+Mx) were more sensitive to Cr(VI) than wild-type AA8 cells and exhibited survival levels comparable with NER-deficient cells (UV-5). Notably, NER-deficient cells (UV-5) that also had BER inhibited by Mx exhibited ∼25% increased sensitivity to Cr(VI), indicating that BER has a distinct role in protecting cells from Cr(VI) toxicity.
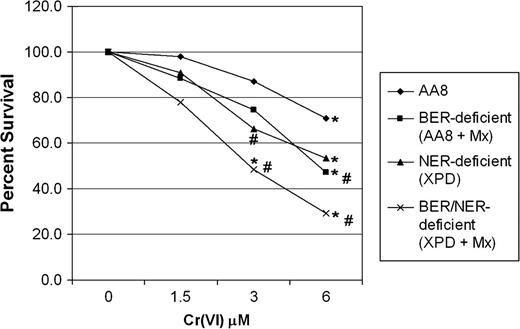
BER plays a role in protecting cells from Cr(VI) toxicity. Repair-proficient (AA8) and repair-deficient (XPD) cells were treated with Cr(VI) ± the BER inhibitor Mx for 24 h. Mx-treated cells were additionally recovered for 24 h prior to plating. Cells were washed and ∼400 cells were plated for clonogenic survival as described in the Materials and Methods. The data are the mean ± SE of at least three independent experiments. *A significant decrease in survival compared with untreated controls as tested for by analysis of variance. #Significant differences between AA8 and AA8 (+Mx), XPD and XPD (+Mx) at specified concentrations.
Cr(VI) mutagenesis is attenuated in BER-inhibited CHO cells
We next sought to determine whether the inhibition of BER by Mx impacted Cr(VI) mutagenesis. As was the case with NER-deficient CHO cells, BER-inhibited CHO cells exhibited an attenuation of Cr-induced HPRT mutagenesis as seen in Figure 4 . At 6 μM Cr(VI), there was a significant decrease in UV-5 (66% reduction) and UV-5 (+Mx) (86% reduction) HPRT mutagenesis relative to AA8 cells.
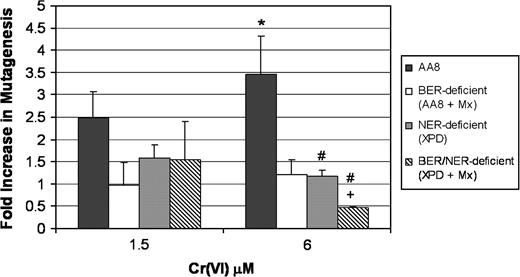
Inhibition of BER by Mx attenuates forward mutagenesis in mammalian cells following Cr(VI) treatment. Repair-proficient (AA8) and repair-deficient (XPD) cells were treated with Cr(VI) for 24 h ± Mx (0.5 mg/ml). Mx cells were recovered for an additional 24 h. Cells were washed and seeded in MEM for recovery (5–7 days). Cells were plated in MEM (plating efficiency) and MEM containing 6-thioguanine (mutant selector) for 5–7 days. The data are the mean ± SE of at least three independent experiments. *A significant increase in AA8 versus untreated controls at the specified concentration as tested for by analysis of variance. #A statistical difference between AA8 and XPD and XPD (+Mx) at the specified doses. +Statistical difference between XPD and XPD (+Mx).
Cr(VI) clastogenesis is attenuated in NER-deficient CHO cells
Cr(VI) is a well-documented clastogen ( 26 , 36 , 37 ). Because Cr(VI) mutagenesis was attenuated in ER-deficient CHO cells, we were interested in determining if Cr(VI) clastogenesis also exhibited a dependence on ER. To test this possibility, we assayed for chromosomal aberrations in repair-proficient and repair-deficient CHO cells. As seen in Figure 5 , Cr(VI) induced a concentration-dependent increase in chromosomal damage from 8 to 32 total events at 1.5 and 6 μM Cr(VI), respectively. Consistent with the HPRT mutagenesis data ( Figure 1A ), Cr(VI) clastogenesis was reduced by 37 and 72% at 1.5 and 6 μM, respectively, in NER-deficient (UV-5) CHO cells. Interestingly, we found that UV-5 cells exhibited a dramatic decrease in the levels of isochromatid lesions (9 events) versus AA8 cells (32 events). Mx by itself is strongly clastogenic in AA8 cells, much more so than Cr(VI). As such, we were unable to test whether inhibition of BER by Mx could modify Cr(VI) clastogenesis.
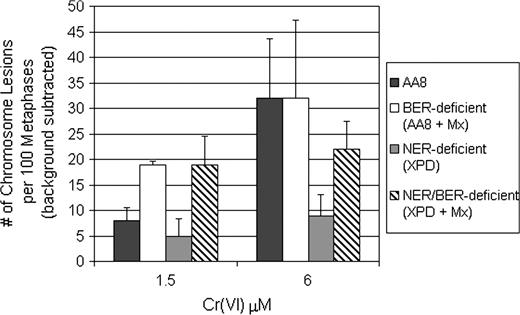
Loss of NER attenuates Cr(VI) clastogenesis. Repair-proficient (AA8) and repair-deficient (XPD) cells were treated with Cr(VI) for 24 h ± Mx (0.5 mg/ml). Mx cells were recovered for an additional 24 h. Metaphases were examined for chromosome damage. The data expressed are the number of total chromosome aberrations per 100 metaphases (background subtracted). The data are mean ± SE of at least three independent experiments.
Discussion
Cr(IV) is a complex genotoxicant and little is known regarding the precise contribution of NER and BER in the protection of cells from Cr(VI) lethality and mutagenesis. Here, we studied the mutation frequency at the HPRT locus in normal and ER-deficient CHO cells. It is a generally accepted dogma that when DNA repair is lost mutagenesis increases. However, the results from this study are the first to indicate that ER is required for chemically induced mutagenesis in mammalian cells. These results were not due to toxicity as equitoxic exposures for NER-proficient and -deficient cells also resulted in attenuated mutagenesis (compare Figures 1A and 3 ). This work has far ranging implications and highlights novel mechanistic aspects of mutagenesis involving the DNA damage response and DNA repair.
Our laboratory has shown previously that NER-deficient CHO cells (UV-41 and UV-5) are moderately sensitive to Cr(VI) toxicity and impeded in the removal of Cr–DNA adducts. This finding was consistent with work by Reynolds et al. ( 19 ) using NER-deficient fibroblasts. In that study, the authors also reported that xeroderma pigmentosum complementation group A cells exhibited hypersensitivity toward Cr mutagenesis. While our results seem to contradict this observation, we contend that the different results can be simply attributed to the system in which mutagenesis was studied.
Specifically, Reynolds et al. employed an extrachromosomally maintained shuttle vector pretreated with Cr and transfected into human cells. After a period of time allowing for repair, they determined mutation frequency by isolating the vector from the fibroblasts, transforming bacteria and measuring the growth of bacteria on selective agar plates. Shuttle vector assays are useful for determining whether specific lesions are mutagenic and identifying the repair mechanisms involved in lesion removal. However, shuttle vectors are not suitable for the study of mutagenesis in chromosomal genes or of DNA replication-arresting lesions, which are selected out because replication-blocked vectors are lost for analysis. One important difference is that shuttle vector studies utilize DNA that has been ‘washed’ of free Cr and is devoid of nucleosomes. Moreover, shuttle vectors are selective for only those lesions that are either easily bypassed by the cellular replication machinery or inherently pose little obstacle to DNA replication. Studies employing shuttle vectors are also less suited for modeling the impact that epigenetic factors play in mutagenesis (i.e. inhibition of DNA repair). We suggest that shuttle vector systems are unable to adequately mimic the net impact of the DNA damage response on mutagenesis at chromosomal loci.
In contrast, we monitored mutagenesis at a chromosomal locus (i.e. HPRT), which is advantageous in that it is impacted by the recognition and repair of chromosomal DNA damage by normal cellular processes in chromatin. This approach detects mutations arising from many mechanisms including replication-arresting lesions. Our results suggest that Cr mutagenesis is highly complex and involves ER when measured using chromosomal sentinel genes. Moreover, it is likely that ER-dependent mutagenesis is tethered to replication fork arrest by Cr–DNA damage.
Cr(VI) reduction leads to lesions that serve as substrates for both NER and BER. For instance, it is well known that Cr(III) participates in the formation of both binary (monoadducts) and ternary (i.e. ICLs, DNA–protein adducts) DNA adducts. Cr(III) also displays an affinity for the phosphodiester backbone which has impeded the structural study of base-specific Cr–DNA adducts. We have shown that Cr(III)–DNA adducts are recognized and processed by bacterial UvrABC endonuclease ( 20 ). In addition to direct metal–DNA interactions, Cr(VI) reduction may also lead to the oxidation of DNA bases which are generally processed by BER. The work by Sugden et al. ( 7 , 8 ) has demonstrated that, in addition to guanine, 8-oxoguanine is further oxidized by a Cr(V)-containing compound. Interestingly, EM9 CHO cells (X-ray cross-complementing group 1 (XRCC1) deficient) are hypersensitive toward clastogenesis by particulate lead chromate, a particulate form of Cr(VI) ( 38 ). Because XRCC1 is involved in the resolution of DNA single-strand breaks (SSBs), which are generated by Cr(VI) exposure ( 39 ), our results collectively suggest that SSBs, but not Cr(VI)-induced base damage, are preclastogenic lesions.
Given that Cr(VI) leads to such a diverse array of genetic lesions, it is not surprising that both NER and BER were found to play a role in protecting cells from Cr(VI) toxicity. What is more intriguing is that both ER pathways are required for mutagenesis. Because several Cr lesions are impediments to DNA replication fork progression, we postulate that ER-dependent mutagenesis is linked to this stimulus. In addition, we cannot rule out the additional possibility that the incision of DNA by ER, during the repair of Cr lesions, and concomitant error-prone ligation and/or repair synthesis might also play a role in this process. Interestingly, we also found that ER is involved in MMS-induced mutagenesis. It is well known that MMS mutagenesis is influenced by methyladenine DNA glycosylase (MAG1). Interestingly, we found that MMS mutagenesis was potentiated in AA8 cells overexpressing XPD ( Figure 2B ). This indicates that XPD is directly involved in ER-dependent mutagenesis following MMS treatment. While the loss of NER created by XPD deletion (i.e. UV-5 cells) attenuates mutagenesis, our data suggest that this specific enzyme plays an even larger role in MMS-induced mutagenesis. Interestingly, we have obtained similar results in yeast indicating that this ER-dependent mutagenesis is a conserved mechanism in eukaryotes (Brooks et al ., submitted for publication). These results indicate that ER-dependent mutagenesis is not unique to Cr(VI) and applies to other genotoxins as well.
Chromosomal instability is a hallmark of lung cancer and is the result of persistent chromosomal damage ( 40 ). The suppression of mutagenesis in the absence, or inhibition, of ER led us to hypothesize that Cr(VI) clastogenesis might also be dependent upon ER. Interestingly, in NER-deficient CHO cells, we observed an attenuation of clastogenesis, suggesting that a common mechanism for NER-dependent mutagenesis and clastogenesis might exist. Given that NER is involved in ICL repair ( 14 ), it is possible that the failure to remove these lesions in NER-deficient cells leads to error-free bypass of the damage [i.e. error-free translesion synthesis (TLS)] leading to decreased mutagenesis. In support of this, we have found that NER-dependent mutagenesis is genetically associated with TLS in yeast (Brooks et al ., submitted for publication). In addition, NER has been implicated in the formation of spontaneous mutations in prokaryotes, which was suggested to involve error-prone TLS ( 41 ). It is also plausible that NER-mediated incision of ICLs, and resulting double-strand breaks, are required for the generation of chromosomal aberrations.
Mutations are associated with a variety of biological processes, including aging and disease progression. With regard to disease progression, specifically neoplastic transformation, mutations are known to activate oncogenes and inactivate tumor suppressor genes, two pathways that work to control cell division. It is generally assumed that DNA repair is antimutagenic and that loss or compromised DNA repair will lead to increased mutagenesis. This is the case in cells exposed to UV irradiation ( 28 , 30 ); however, the results from this study indicate that this dogma does not always apply. These results are consistent with earlier work, which reported on the insensitivity of NER-deficient bacteria ( 42 ) and yeast ( 43–45 ) to mutagenesis. In an attempt to better understand the mechanisms of Cr(VI) mutagenesis, we have, for the first time, made the observation that both NER and BER are actually required for mutagenesis following treatment with chemical mutagens. This suggests that, in some cases, cells may utilize DNA repair pathways as survival mechanisms at the expense of decreased fidelity. Future work will focus on elucidating the molecular mechanisms involved in ER-dependent mutagenesis and the relative contribution that TLS and replication stress signaling plays in this process.
Funding
This work was supported by a grant from the National Institutes of Health (ES005304).
Abbreviations
- AP
apurinic/apyrimidinic
- BER
base excision repair
- CHO
Chinese hamster ovary
- Cr(III)
trivalent chromium
- Cr(VI)
hexavalent chromium
- ER
excision repair
- ICL
interstrand cross-link
- MEM
minimal Eagle’s medium
- MMS
methylmethane sulfonate
- Mx
methoxyamine hydrochloride
- NER
nucleotide excision repair
- TLS
translesion synthesis
- UV
ultraviolet
- XP
xeroderma pigmentosum
- XPD
xeroderma pigmentosum complementation group D
- XPF
xeroderma pigmentosum complementation group F
This work was conducted in partial fulfillment of the requirements for the PhD degree in Molecular and Cellular Oncology, Columbian Graduate School of Arts and Sciences, George Washington University.
Conflict of Interest Statement: None declared.
References
Author notes
These authors contributed equally to this work.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}